Uracil as an Alternative
to 5-Fluorocytosine
in Addressable Protein Targeting
by
John A. Wendel and Steven S. Smith
City of Hope
1500 E. Duarte Rd.
Duarte, CA 91010-3000
[email protected]
This is a draft paper
for a talk at the
Fifth
Foresight Conference on Molecular Nanotechnology.
The final version has been submitted
for publication in the special Conference issue of Nanotechnology.
This page uses the HTML <sup> and <sub>
conventions for superscripts and subscripts. If "103"
looks the same as "103" then your browser does not
support superscripts. If "xi" looks the
same as "xi" then your browser does not support
subscripts. Failure to support superscripts or subscripts can
lead to confusion in the following text, particularly in
interpreting exponents.
Abstract
Bacterial DNA methyltransferases offer an approach to
addressable protein targeting in macromolecular assembly that
permits the construction of ordered arrays of functional proteins
or peptides. This approach uses the natural recognition
specificity of the bacterial DNA cytosine methyltransferase to
target fusion proteins to preselected sites on a DNA scaffold
through the formation of a stable covalent attachment to a
mechanism-based methyltransferase inhibitor. Addressable targets
on DNA molecules can readily be created with 5FdC* at the enzyme
recognition site. This is because the substitution of the
targeted pyrimidine ring with fluorine at C5 generates a kinetic
barrier to the methyltransfer step in the reaction and a
thermodynamic barrier to the beta-elimination step that stall the
enzyme after it forms a covalent bond with DNA. In this report we
have studied the capacity of dU to substitute for 5FdC in
addressable targeting. When dU is used in place of 5FdC as the
base targeted by the enzyme for nucleophilic attack, it forms a
mispair with dG. Like 5FdC, dU creates a kinetic barrier to
completion of the reaction causing the enzyme to stall. Unlike
5FdC, dU does not create a thermodynamic barrier to
beta-elimination and release after methyltransfer, however, the
mispaired state of the dU in the dG:dU basepair appears to create
a transition-state analog that cannot be easily released by the
enzyme. The data show that dU can be used to substitute for 5FdC
in the production of ordered nucleoprotein-based macromolecular
assemblies.
|
| * Abbreviations Used: FdC, 5-fluorodeoxycytidine; dU,
deoxyuridine; dG, deoxyguanosine; dC, deoxycytidine; mdC,
5-methyldeoxycytidine; mdU,
5-methyldeoxyuracil = dT, deoxythymidine. |
Introduction
One of the problems in producing nanoscale assemblies and
devices has been developing technologies for the control of
position ( Eigler, et al., 1990, Merkle, 1993 ). Living systems routinely
produce nanoscale biostructures that take advantage of precise
spatial arrangements.The assembly of higher order biostructures
in living systems generally depends on the exquisitely selective
interactions between proteins, between nucleic acids, and between
proteins and nucleic acids that have evolved over millenia.
Chromatin, the complex aggregate of proteins and nucleic acids
that forms the fundamenal material in chromosomes, is composed of
precisely ordered functional proteins and DNA. In general,
protein-DNA interactions in chromatin are reversible, so they can
disassemble and reassemble during the cyclic processes that are
required in cellular reproduction. However, some of the spatial
arrangements between proteins on DNA appear to be set down as
patterned biostructures that are copied precisely from one cell
to the next and can remain intact for many years.
Considerable research in this area of molecular biology has
focused on how such patterns are established and maintained since
this is not only the central question of developmental biology,
it is a key question in biological research on aging and cancer.
One aspect of these studies has been research on the function of
DNA methyltransferases and their biological role in this process.
This research suggests that these molecules have been designed by
evolution and natural selection to stabilize biostructures in
chromatin by physically participating in the assembly of these
structures (Smith, 1998). Whether or not
this interpretation of the function of methyltransferases in
biology turns out to be correct remains to been seen. However,
the capacity of these enzymes for positionally controlled
self-assembly can easily be utilized in the production of stable
ordered arrays of functional proteins on DNA in vitro (Smith, et al., 1995, Smith,
et al., 1997 and Smith, et al.,
1997a).
Control of positioning for molecular components is achieved
through the addressing capacity of these enzymes that is rooted
in their sequence specificity and their mechanism of action. Of
the more than 40 different bacterial methyltransferases and the
several eukaryotic methyltransferases that have been cloned and
carefully studied, all have well characterized DNA sequence
specificities (McClelland, M. & Nelson, M., 1988). A subset of these, the
cytosine-5 methyltransferases, form abortive covalent complexes
between an active-site cysteine and 5-fluorocytosine in their DNA
recognition sequences (Osterman, et al.,
1988, Chen, et al., 1991, Smith, et al., 1992). This permits the
cytosine methyltransferases to be converted into targeting
devices by molecular cloning techniques that produce fusion
proteins containing a functional enzyme, because a
methyltransferase with a given sequence specificity will serve to
uniquely target that address. For example, the cytosine
methyltransferases like M·HhaI and M·MspI have
distinct recognition specificities. When 5-fluorocytosine (F) is
placed at the targeted cytosine in each recognition sequence
(GFGC for M·HhaI and FCGG for M·MspI) the first
recognition site becomes a unique address for M·HhaI and
the second recognition site becomes a unique address (Figure 1) for M·MspI (Smith,
et al., 1997, Smith, et al.,
1997a). This permits precise control over the positioning of
proteins or peptides fused to the methyltransferases (Smith, 1997a).
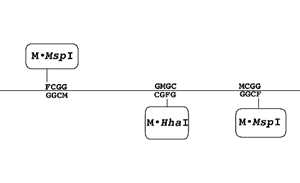
Larger version (22K)
Figure 1. Schematic of a three factor linear
addressing system. The recognition sites for M·HhaI and M·MspI
are placed at preselected points along a linear duplex DNA
molecule. Methylation sites within the recognition sequence
that are targeted by the methyltransferase are replaced with
5FdC (F). Attack of the FdC moiety by the appropriate enzyme
produces a covalently linked complex between the enzyme and
its recognition site. (M): mdC.
The availability of DNA scaffolds of almost any topology from
the connectivity of a cube to that of a truncated octahedron (Seeman, 1993, Zhang and Seeman, 1992; Zhang and Seeman, 1994) now makes it possible to
envision such scaffolds decorated with a variety of functional
peptides or proteins in precisely controlled ways. In this report
we have studied the utility of dU in the trapping
methyltransferases at addressable sites in DNA.
Materials and Methods
DNA synthesis: Oligodeoxynucleotides were synthesized
on a Cyclone Plus DNA Synthesizer (Millipore, Marlborough, MA)
using standard phosphoramidite chemistry. Precursor
phosphoramidites were purchased from PerSeptive Biosystems
(Farmington, MA) or from Glenn Research (Sterling, VA). They were
then purified using Oligo-Pure cartridges (Hamilton, Reno, NV)
according to the manufacturer's protocol. This was followed by 32P
end-labelling, as previously described (Smith,
et al. 1991).
Mobility Shift Assays: Duplexes were formed by
combining equimolar (20 µM) amounts of complementary strands in
annealing buffer (10 mM Tris-HCl pH 7.4, 1 mM EDTA, and 100 mM
NaCl) then treating with 95°C for 5 minutes and 50°C for 60
minutes. The samples were allowed to cool to room temperature for
10 min. and were then stored on ice until needed.
Methyltransferase Purification: M·HhaI was
obtained via purification from E. coli RR1 containing the
pSP72 plasmid (Promega, Madison, WI) carrying the entire HhaI
methyltransferase gene ( Smith, et al.
, 1997a). Transcription was from the endogenous hsdM promoter.
Formation of Assemblies: M·HhaI was incubated
with either a 30mer containing a single recognition site with dU
(bold) at the target site:
5'TCACCAGAT GCCG GUGC GTGACCTGTAGTT3'
3'AGTGGTCTA CGGC CGMC CACTGGACATCAA5'
or a 60mer containing two recognition sites with dU (bold) at
each target site :
5'TCACCAGAT GUGC TGTAGGTCGT GCTACCTGGT TCCACCAGAT GUGC
GTGACCTGTAGTT3'
3'AGTGGTCTA CGMG ACATCCAGCA CGATGGACCA AGGTGGTCTA CGMG CACTGGACATCAA5'.
M:mdC.
The reaction took place overnight at 37°C in a buffer containing 50 mM Tris-HCl pH 7.5, 10 mM EDTA, 5 mM 2-mercaptoethanol, 80 µM S-adenosyl-[L]-methionine (New England Biolabs, Beverly,MA), and 4 µM oligodeoxynucleotide duplex. The reaction mixture was separated on a 6-20% non-denaturing gradient polyacrylamide gel containing a 4% stacking gel (Smith, et al., 1997). The gel was then exposed directly to X-ray film (Kodak, Rochester, NY) for autoradiography. 32P end-labelled fragments of TaqI-digested phiX174 RFII (New England Biolabs, Beverly, MA) were used as molecular length markers for DNA.
Ab initio Methods: We performed ab
initio calculations on a number of variants of cytosine and
uracil structures which we believe accurately model key
intermediates in the proposed reaction pathway. Ab initio
geometries were calculated at the Hartree-Fock level of theory
using the STO-3G basis set with SPARTAN 4.0 (Wavefunction,
Irvine, CA) running on a network of Silicon Graphics
workstations. Single-point ab initio orbital energies
calculated with the 6-31G* basis set were used to construct
electron density surfaces with color maps of frontier orbital
values. Blue indicates a high value for the orbital, and red
indicates a low value.
Enzyme Activity Assay: Activity was determined using
trichloroacetic acid-precipitable radioactivity retained on glass
fiber filters as described (Smith et al., 1992). The M·HhaI
assay was assayed in a buffer containing 50 mM Tris-HCl (pH 7.5),
10 mM EDTA, 5 mM 2-mercaptoethanol, 6µM S-adenosyl-L-[3H-methyl]methionine
15 Ci/mmole (Amersham, Arlington Heights, IL) and 5 µM
oligodeoxynucleotide duplex 30-mer for one hour at 37°C.
Molecular Modeling: Molecular models of the assembly
formed between the 60mer containing dU at the target site were
constructed in BIOGRAF 3.21 (Molecular Simulations, San Diego,
CA). The initial conformation of M·HhaI was that
determined from the crystalline protein complexed with
5-fluorocytosine at its target site (Klimasauskas,
et al., 1994). In the DNA, the four nucleotides at the
target site were taken from the crystal structure data, except
that 5FdC was converted directly to dU. The remainder of the
duplex 60mer was constructed using the software's DNA builder.
The structure was minimized in molecular mechanics to 0.1
(kcal/mole)/Å, and rendered using standard visualization tools
available in the program. The model assumes linear DNA outside of
the target sequences with the pitch of 10.0 bp/turn (i.e.,
the helical twist of 36.0°/bp derived from fiber diffraction).
With this assumption, the C termini of the two enzymes assume the
180° dihedral angle, measured down the helical axis, shown in
the model. Although the DNA is not bent by the enzyme, unwinding
of the helix within the DNA binding site has been observed in the
M·HhaI DNA complex. This unwinding could result in a
twist angle of 31.6°/bp for those base pairs in the binding
site. Since it is also possible that the twist angles for the DNA
outside the binding sites could be as low as 34.3°/bp based on
solution measurements, the true dihedral angle could be anywhere
from 80° to 180° .
Results and Discussion
In the dC reaction catalyzed by DNA
(cytosine-5)-methyltransferases (Figure 2),
binding of the DNA substrate initiates a series of conformational
changes that result in the opening of a salt bridge present
between an arginine and a glutamic acid in the apoenzyme. Binding
of the components of the salt bridge to the deoxycytosine moiety
in the DNA substrate is facilitated by the capture of a proton in
a hydrogen bond formed between an oxygen on glutamic acid and N3
of the targeted cytosine. Nucleophilic attack at the C6 position
of the cytosine moiety by a cysteine residue at the active site
rapidly produces a resonance-stabilized carbanion that is
activated to attack a methyl group present on bound S-Adenosylmethionine
co-factor, through C5 of cytosine. The resulting dihydrocytosine
intermediate rapidly undergoes beta-elimination to produce free
enzyme, S-adenosylhomocysteine and DNA in which the
targeted cytosine moiety has been converted to 5-methylcytosine.
Figure 2: Attack of Deoxycytosine by DNA (Cytosine-5)Methyltransferases. Top: Chemical Depiction of Intermediates. In this model of the chemical reaction, the break-up of the glutamic acid-arginine salt bridge is stabilized by the capture of a proton poised between the ionized carboxyl oxygen of the glutamic acid and N3 of cytosine. This activates the ring for nucleophilic attack by a cysteine residue also at the active site. In the model shown here, the captured proton is assumed to be derived from the cysteine sulfhydryl increasing its nucleophilicity. Once nucleophilic attack occurs, the resonance-stabilized carbanion attacks the methyl group on S-adenosylmethionine to generate a dihydrocytosine intermediate. Resonance stabilization is enhanced by the proximity of the positively charged guanidinium group on arginine which forms a hydrogen bond with O2 of the intermediate, and is in a position to ligate negative charge on O2 generated through resonance. This intermediate undergoes beta-elimination to generate a 5-methylcytosine product and active enzyme. A tightly bound water molecule on the enzyme surface appears to be the proton acceptor in the beta-elimination step. Bottom: Quantum Chemical Depiction of Models of the Intermediates. Ab initio geometries were calculated at the Hartree-Fock level of theory using the STO-3G basis set with Spartan 4.0 (Wavefunction, Irvine, CA) running on a network of Silicon Graphics workstations. Single-point ab initio orbital energies calculated with the 6-31G* basis set were used to construct electron density surfaces with color maps of frontier orbital values. Blue indicates a high value for the orbital and red indicates a low value. High values for the Lowest Unoccupied Molecular Orbital (LUMO) are seen over C6 and C4 for 1-methylcytosine, indicating that these are potential acceptor sites for nucleophilic attack.The 1-methyl-6-sulfhydryl-cytosine carbanion was used to model the intermediate formed by the enzyme after nucleophilic attack at C6. High values of the Highest Occupied Molecular Orbital are confined to C5. This highly reactive orbital is then poised for attack on the methyl of S-adenosylmethionine.
When FdC is placed at the target site (Figure
3) the reaction proceeds in the same fashion. However, the
methyltransfer step is slowed dramatically by the presence of the
FdC moiety (see below) and the beta-elimination step is blocked
by the presence of the fluorine atom at C5.
Figure 3: Attack of 5-Fluorodeoxycytosine by DNA (Cytosine-5)Methyltransferases.Top: Chemical Depiction of Intermediates. Enzymatic attack on 5FdC proceeds as in Figure 1 for attack on cytosine. The resonance-stabilized carbanion of 5FdC can attack the methyl group on S-adenosylmethionine to generate a dihydrocytosine intermediate at a very slow rate. In the dC reaction, this intermediate would undergo beta-elimination to generate a mdC product and active enzyme. 5FdC, blocks beta-elimination because the fluorine at C5 cannot be abstracted. Bottom: Quantum Chemical Depiction of Models of the Intermediates. Ab initio geometries were calculated at the Hartree-Fock level of theory using the STO-3G basis set with Spartan 4.0 (Wavefunction, Irvine, CA) running on a network of Silicon Graphics workstations. Single-point ab initio orbital energies calculated with the 6-31G* basis set were used to construct electron density surfaces with color maps of frontier orbital values. Blue indicates a high value for the orbital and red indicates a low value. High values for the Lowest Unoccupied Molecular Orbital (LUMO) are seen over C6 and C4 in the 1-methyl-5-fluorocytosine used to model the intermediate, indicating that these are potential acceptor sites for nucleophilic attack as with cytosine. The 1-methyl-6-sulfhydryl-carbanion derivative of 5-fluorocytosine was used to model the intermediate formed by the enzyme after nucleophilic attack at C6. High values of the Highest Occupied Molecular Orbital are again confined to C5. This highly reactive orbital is then poised for attack on the methyl of S-adenosylmethionine.
When dU is placed at the target site (Figure 4),
the progress of the reaction is disturbed in several ways by the
presence of the mispaired target base. First, the reorientation
of the salt bridge is not expected to draw a proton into a
hydrogen bond between O4 and the oxygen until nucleophic attack
occurs at C6. The formation of the resonance stabilized O4 enol
dramatically reduces the capacity of the system to attack the
methyl on S-Adenosylmethionine. Like the reaction with
FdC, the reaction with dU is slowed by the presence of the
additional electronegative atom on the ring (i.e. O4).
However, unlike the reaction with FdC, the beta-elimination step
is not blocked.
Figure 4. Attack of Deoxyuracil by DNA (Cytosine-5) Methyltransferases.Top: Chemical Depiction of Intermediates. The initial nucleophilic attack of uracil by the enzyme is not favored because the resulting intemediate must develop negative charge on O4 as the negatively charged carboxyl group on glutamic acid forms a hydrogen bond with N3. Decay of the system through protonation of O4 (perhaps by the cysteine sulfhydryl on the enzyme) can produce a resonance stabililized carbanion much like that produced from 5-fluorocytosine. Attack of the methyl group on S-adenosyl-[L]-methionine by the carbanion followed by beta-elimination would produce 5-methyluracil (thymidine). Bottom:Quantum Chemical Depiction of Models of the Intermediates. High values for the Lowest Unoccupied Molecular Orbital (LUMO) are seen over C6 and C4 1-methyl-deoxyuracil, indicating that these are potential acceptor sites for nucleophilic attack as with cytosine. The 1-methyl-6-sulfhydryl-4-ol-carbanion derivative of deoxyuracil was used to model the intermediate formed by the enzyme after nucleophilic attack at C6. High values of the Highest Occupied Molecular Orbital are again confined to C5. Once methyl-transfer takes place, the high values of the Highest Occupied Molecular Orbital (HOMO) move to the sulfur atom in (1-methyl-6-sulfhydryl-5-methyl-deoxyuracil).
The nature of the chemical barrier to completion of the
reaction is best measured by the energy of the highest occupied
orbital of the carbanion (Figure 5).
Figure 5. Electronic Trapping of the Methyltransferase. The observed rates of enzyme catalyzed methyltransfer observed for FdC and dU are plotted relative to that observed for dC as a function of the calculated ab initio energy of the Highest Occupied Molecular Orbital E(HOMO) of the enzyme activated intermediate.
For dC this corresponds to -0.11278 hartree, in FdC this is
reduced to -0.11813, and for the attacking dU enol it is reduced
to -0.11939. At 4 µM oligodeoxynucleotide, the rate of
methyltransfer is slowed by 70 to 200 fold depending on the
oligodeoxynucleotide employed. Clearly both dU and FdC are
powerful mechanism-based inhibitors of the methyltransferase
reaction. Even so, the existence of a measurable rate of
methyltransfer (2.8 fmole/min) to dU under these conditions
suggests that the complexes should decay slowly. Since the
complexes formed with dU appear to have a stability similar to
the stability of complexes formed by FdC (Klimasauskas
and Roberts, 1995, Yang,
et al., 1995) there must be other factors that contribute
to the stability.
One possibility is that there is a significant kinetic barrier
established by the requirement for removal of the stabilizing
proton at O4. In fact the system could stall if the relatively
stable enol form of the intermediate were to form after
methyltransfer. On the other hand, recent work with the human
enzyme shows that it can stall at dC when the cytosine is
mispaired. That inhibition stems from the fundamental catalytic
imperative in enzymology: that an enzyme have a higher affinity
for the transition state of a reaction than it has for either its
reactants or its products (Haldane, 1930, Pauling, 1948).
For the dC methyltransferase reaction, the formation of the
dihydrocytosine intermediate requires that the base unstack from
the helix (Baker, et al., 1988)
because the aromaticity of the ring is destroyed by the
introduction of sp3 carbons at C5 and C6 at the same
time that the geometry of the ring is changed from the planar
conformation required for normal stacking in DNA to the
non-planar conformation required for catalysis to proceed. This
prediction has been confirmed by numerous experiments (Baker, et al., 1988, Smith,
et al., 1991, Laayoun, et al.,
1994) and was essentially proven by crystallographic work on the
structure of the FdC complex formed between M·HhaI and
FdC containing DNA (Klimasauskas, et al.,
1994), which showed that the target base is rotated into a
completely extrahelical conformation in order to reach the
catalytic site on the enzyme surface.
Thus, any weakly stacked or extrahelical cytosine is a
transition state analog for the catalysis (Smith,
1994) for which the enzyme is expected to have a very high
affinity. The mispaired base appears to present a structural trap
to the enzyme since it is locked in a structure resembling the
transition state by the structure of the mispaired DNA substrate
itself. In effect, the enzyme is expected to rapidly bind and
attack the substrate but it is not expected to be able to release
the methylated product (if it forms) because it retains a high
affinity for the unstacked transtion-state analog. The
displacement of the mispaired 5-methyldeoxyuracil product (mdU)
in the dG:mdU mispair relative to the normally paired
dG:mdC product is depicted in Figure 6.
Figure 6. Structural Trapping of the Methyltransferase. The crystal structures of the dG:dC substrate for the methyltransferase (Drew et al., 1981) and that of its normal dG:mdC product (Timsit and Moras, 1995) are compared with the structure of the mispaired dG:mdU product (Hunter, et al., 1987) expected after methylation of dU. Notice the strong displacement of the mdU moiety into the major groove of the helix, and the weaker displacement of the dG moiety into the minor groove necessitated by the abberrant hydrogen bonding scheme in the mispair.
Each of these considerations suggests that methyltransferases
stalled at dU should be as stable as methyltransferases bound at
FdC. The experiment depicted in Figure 7
shows that a stable assembly can be formed with a DNA 60mer
containing two dU residues at targeted sites in the 60mer spaced
35 nt apart and M·HhaI. The yield and stability of the
complex are comparable to that of the complex formed between M·HhaI
and this same 60mer sequence containing FdC at the sites occupied
by the dU in this oligodeoxynucleotide duplex (Smith,
et al., 1997). A molecular model of the biostructure
formed with dU is depicted in Figure 8.
Figure 7. Formation of Enzyme-DNA Complexes with dU-Containing Oligodeoxynucleotides. (a) A standard curve of M·MspI and M·HhaI molecules complexed with duplex 64mers or duplex 60mers containing an FdC residue at the enzymatic target site. Bacterial enzymes linked to DNAs of known lengths (Smith, et al., 1997), were constructed as described in Materials and Methods. (b) Electrophoretic separation of complexes formed between M·HhaI and dU-containing oligonucleotides. Markers in (a) identify the complexes as diagramed in the center of the figure. Markers to the right of panel (b) show the positions of duplex DNA molecules of the indicated lengths.

Larger version (80K)
Figure 8. Molecular Model of the Assembly Formed between a Duplex DNA 60mer Containing dU Moieties at Target Sites Spaced 35nt Apart. The model shown was constructed and rendered as described in Materials and Methods. In this rendering the C-termini of the two M·HhaI molecules (white) lie roughly on a vertical line that bisects the image.
Conclusions
In this report we have used a simple technology demonstration
system to show that both FdC and dU can be used to address
methyltransferases to preselected sites on DNA. Our data suggest
that other mechanism-based inhibitors of these enzymes will also
be useful in this application, and when coupled with previous
work, suggests that a variety of useful biostructures and devices
can be constructed with this technology.
*Supported by grant N00014-94-1-1116 from the Office of Naval Research
References
- Baker, D.J.; Kan, J.L.C.; &
Smith, S.S. (1988) Gene 74, pages 207-210.
Recognition of Structural Perturbations in DNA by Human
DNA (Cytosine-5)Methyltransferase.
- Chen, L.; Chen, L.; MacMillan, A.M.;
Chang, W.; Ezaz-Nikpay, K.; Lane, W.S. & Verdine,
G.L. (1991) Biochemistry 30, pages 11018-11025.
Direct Identification of the Active-Site Nucleophile in a
DNA (Cytosine-5)Methyltransferase.
- Drew, H.R.; Wing, R.M.; Takano, T.;
Broka, C.; Tanaka, S.; Itakura, K.; & Dickerson, R.E.
(1981) Proc. Natl. Acad. Sci. USA 78, pages
2179-2183. Structure of a B-DNA dodecamer: conformation
and dynamics.
- Eigler, D.M.; & Schweizer, E.K.
(1990) Nature 344, pages 524-526. Positioning
Single Atoms with a Scanning Tunnelling Microscope.
- Haldane, J.B.S. (1930)
"Enzymes", Longmans Green & Co. Ltd.; Great
Britain, Reprinted by MIT press (1965).
- Hunter W.N.; Brown, T.; Kneale, G.;
Anand, N.N.; Rabinovich, D.; Kennard, O. (1987) J. Biol.
Chem. 262 9962-9970. The Structure of
Guanosine-thymidine Mismatches in B-DNA at 2.5-A
Resolution.
- Klimasaukas, S.; Kumar, S.; Roberts,
R.J. & Cheng, X. (1994) Cell 76, pages
357-369. HhaI Methyltransferase Flips Its Target Base Out
of the DNA Helix.
- Klimasaukas, S.; & Roberts, R.J.
(1995) Nucleic Acids Res. 23, pages 1388-1395.
M·HhaI Binds Tightly to Substrates Containing Mismatches
at the Target Base.
- Laayoun, A.; Baker, D.J.; Riley, J.
& Smith, S.S. (1994) Gene 150, pages 195-196.
The Response of M·HpaII to Heteroduplexes.
- McClelland, M. & Nelson, M.
(1988) Gene 74, pages 291-304. The Effect of
Site-Specific Methylation on Restriction Endonucleases
and DNA Modification Methyltransferases -A Review.
- Merkle, R.C. (1993) Chemical Design
Automation News, 8, Nos. 9 & 10, September/October.
Molecular Manufacturing: Adding Positional Control to
Chemical Synthesis.
- Osterman, D.G.; DePillis, G.D.; Wu,
J.C.; Matsuda, A. &, Santi, D.V. (1988) Biochemistry 27,
pages 5204-5210. 5-Fluorocytosine in DNA is a
Mechanism-Based Inhibitor of HhaI Methylase.
- Pauling, L.; (1948) American
Scientist 36, pages 50-58. Chemical Achievement
and Hope for the Future.
- Smith, S.S.; (1994) Prog. in Nucleic
Acids Res. and Molec. Biol. 49, 65-111, Waldo E.
Cohn and Kivie Moldave, Eds., Academic Press, N.Y., N.Y.
Biological Implications of the Mechanism of Action of
Human DNA Methyltransferases.
- Smith, S.S.; (1995) In: Biological
and Biomedical Science and Technology Division 1995
Programs, ed. E. Eisenstadt, (U.S. Navy Publication ONR
34196-3), pages 161-162. Nucleoprotein-Based Nanoscale
Fabrication.
- Smith, S.S.; (1998) Int. J. Molec.
Med., in press. Stalling Of DNA Methyltransferase in
Chromosome Stability and Chromosome Remodelling.
- Smith, S.S.; Kan, J.L.C.; Baker,
D.J.; Kaplan, B.E. & Dembek, P. (1991) J. Mol. Biol. 217,
pages 39-51. Recognition of Unusual DNA Structures by
Human DNA (Cytosine-5)Methyltransferase.
- Smith, S.S.;. Kaplan, B. E., Sowers,
L. C. & Newman, E. M. (1992) Proc. Natl. Acad. Sci.,
U.S.A. 89, 4744-4748. Mechanism of Human
Methyl-Directed DNA Methyltransferase and the Fidelity of
Cytosine Methylation.
- Smith, S.S.; Niu, L.; Baker, D.J.;
Wendel, J.A.; Kane, S.E. & Joy, D.S. (1997) Miami
Biotechnology Short Reports, Vol. 8. F.Ahmad et
al., eds. IRL at Oxford University Press, page 13.
Nanoscale Addressing in Macromolecular Assembly.
- Smith, S.S.; Niu, L; Baker, D.J.;
Wendel, J.A.; Kane, S.E. & Joy, D.S. (1997a). Proc.
Natl. Acad. Sci. U.S.A. 94, pages 2162-2167.
Nucleoprotein Based Nanoscale Assembly.
- Seeman, N.C. (1991) DNA and Cell
Biology 10, pages 475-486.Construction of
Three-Dimensional Stick-Figures from Branched DNA.
- Yang, A.S.; Shen, J-.C.; Zingg,
J-.M.; Mi, S. & Jones, P.A. (1995) Nucleic Acids Res.
23, pages 1380-1387. HhaI and HpaII DNA
Methyltransferases Bind DNA Mismatches, MethylateUracil
and Block DNA Repair.
- Zhang, Y. & Seeman, N.C. (1992)
J. Am. Chem. Soc. 114, pages 2656-2663. A
Solid-Support Methodology for the Construction of
Geometrical Objects from DNA.
- Zhang, Y. & Seeman, N.C. (1994)
J. Am. Chem. Soc. 116, 1661-1669. Construction of
a DNA-Truncated Octahedron.
- Timsit,Y.& Moras D. (1995) J.
Mol. Biol. 251, pages 629-647. Self-fitting and
Self-modifying Properties of the B-DNA Molecule.
|
| Last Modified by authors:
02:01am PDT, October 04, 1997 |
|