Electronic Structure of Thiolate Self-Assembled Monolayers on Cu(111) and Au(111) substrates
aDepartment of Physics, Brookhaven National Laboratory,
Upton, New York 11973 USA
bChemistry Department, Brookhaven National Laboratory,
Upton, New York 11973 USA
cDepartment of Physics, University of Wisconsin-Milwaukee,
Milwaukee, WI 53201 USA
This is an abstract
for a presentation given at the
10th
Foresight Conference on Molecular Nanotechnology
Recently there has been increased interest in the possibility of using organic molecules as possible electronic components in nanoscale devices. One of the most important issues to be addressed is the role of molecule-surface interactions. Density Functional Theory (DFT) calculations can provide important information regarding the electronic structure of these type of systems, but up to now generally have used either a finite cluster of metal atoms with an attached molecule or a repeated supercell geometry. Both approaches suffer from various shortcomings, either a poor representation of the delocalized metallic states (clusters) or unphysical cell-cell interactions (supercells). The Full-potential Linear Augmented Plane Wave (FLAPW) in the free-standing film geometry avoids these shortcomings. In this approach, the substrate is modeled by a N-layer (typically N~9-25) two-dimensional film and the region beyond the substrate and adsorbate treated as a true "vacuum" region with the proper boundary conditions at infinity.
To demonstrate the feasibility of determining the interactions between organic molecules and metallic surfaces, we calculate the properties of a nine layer Cu(111) film with an attached 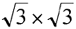 R30 thiolate (SC6H5) self-assembled monolayer (SAM). A recent two photon experiment [1] reports a work function shift from 4.9 eV for the clean Cu surface to 3.7 eV with the adsorbed SAM. Additionally, two states were resolved, one below the vacuum level by 0.4 eV and the other 2.6 eV above. The DFT results predicts a larger the work function reduction due to the SAM formation, which is attributed to a small area 16.9 Å2 per molecule. For the Au surface the area is 21.6 Å2, which corresponds to the optimal packing. We report the geometry and electronic structure necessary for the detailed interpretation of the experiments. R30 thiolate (SC6H5) self-assembled monolayer (SAM). A recent two photon experiment [1] reports a work function shift from 4.9 eV for the clean Cu surface to 3.7 eV with the adsorbed SAM. Additionally, two states were resolved, one below the vacuum level by 0.4 eV and the other 2.6 eV above. The DFT results predicts a larger the work function reduction due to the SAM formation, which is attributed to a small area 16.9 Å2 per molecule. For the Au surface the area is 21.6 Å2, which corresponds to the optimal packing. We report the geometry and electronic structure necessary for the detailed interpretation of the experiments.
A crucial advantage of the our FLAPW code is the ability to self-consistently include the effects of an external applied electric field, which will allow us to more realistically model the properties of the molecule/surface interface corresponding to nanoelectronic devices.
Reference
- T. Vondrak, H. Wang, P. Winget, C. J. Cramer, and X.-Y. Zhu, J. Am. Chem. Soc. 122, 4700 (2000).
Abstract in Microsoft Word® format 17,734 bytes
*Corresponding Address:
Vasili Perebeinos
Department of Physics, Brookhaven National Laboratory
20 Pennsylvania Street, Upton, New York 11973 USA
Phone: (631)-344-3225 Fax: (631)-344-2918
Email: [email protected]
Web: http://cmth.phy.bnl.gov/~perebein/
|