Non-Covalent Assembly of Functional Surfaces
Department of Chemistry and Biochemistry, Worcester Polytechnic Institute,
Worcester, MA 01609 USA
This is an abstract
for a presentation given at the
11th
Foresight Conference on Molecular Nanotechnology
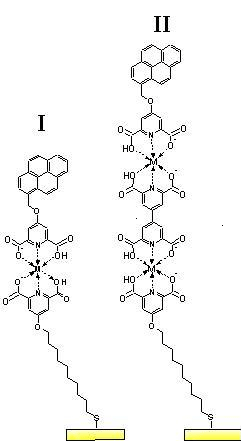
Non-covalent metal-ligand binding interactions have been used to fabricate multilayered thin films that generate photocurrent upon exposure to light. Films have been constructed by the sequential deposition of two components on gold that serve as a template for further deposition. Thus, 4-[(10-mercaptodecyl)oxy]pyridine-2,6-dicarboxylic acid is deposited first and forms a highly ordered and electrically insulating self assembled monolayer (SAM) on the gold surface. Cu(II) ions are subsequently deposited and complex with the two carboxylate groups and the nitrogen atom on the pyridine head group. Several different films have been constructed from this initial template. For example, Film I consists of the template capped by another dicarboxypyridine group tethered to a light absorbing pyrene chromophore. Film II is capped by a tetracarboxydipyridyl group, a subsequent layer of Cu(II) ions and finally by the same pyrene group. Incorporation of these films into an electrochemical cell as the working electrode and irradiation produces photocurrent with an efficiency of roughly 2%. Similar results were achieved using different transition metal ions, including Co(II) and Fe(III). In addition, films consisting of multiple layers of metal ions and pyridine groups (up to 5 layers of metal) yield substantial photocurrent upon irradiation.
This non-covalent approach to the fabrication of multilayered films provides a simpler method for the construction of a wide variety of molecular scale devices incorporating multiple components, than large covalently linked molecules.
Abstract in Microsoft Word® format 53,574 bytes
*Corresponding Address:
W. Grant McGimpsey
Department of Chemistry and Biochemistry, Worcester Polytechnic Institute,
100 Institute Road
Worcester, MA 01609 USA
Phone: 508-831-5486 Fax: 508-831-5933
Email: [email protected]
|