Ab-Initio simulation of molecular devices
M. Di Ventra*, a, S.T. Pantelidesa, and N.D.
Langb
aDepartment of Physics and Astronomy,
Vanderbilt University, Nashville, Tennessee 37235
bIBM Thomas J. Watson Research Center, Yorktown Heights, New York 10598
This is an abstract
for a presentation given at the
Sixth
Foresight Conference on Molecular Nanotechnology.
There will be a link from here to the full article when it is
available on the web.
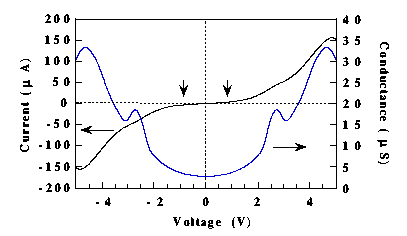
We report the first parameter-free first-principles calculation of the I-V characteristics of a molecular device. We investigated the benzene-1,4-dithiolate molecule, the same molecule for which the I-V characteristics have been measured by Reed et al. [1]. The results are shown in Fig. 1. The shape of the curve is similar to the experimental one, but the absolute magnitude of the current is significantly different, namely the theoretical resistance of the molecule is significantly lower than what is measured.
| |
Fig. 1. Conductance of a benzene-1,4-dithiolate molecule as function of the external bias. The vertical arrows indicate the region where the conductance is practically constant, in agreement with experiment. |
|
|
Since the numerical convergences have been thoroughly tested and the theory has no adjustable parameters, we attribute the discrepancy between theory and experiment to contact resistances. As a check, we inserted an H atom at the contacts and the absolute magnitude of the current increased by an order of magnitude. The presence of the H atoms at both contacts simply opens up additional conduction channels because of the strong C-H bond. Rotation of the benzene ring with respect to the S-H contacts, instead, decreases the conductance to values comparable to the case without H. Calculations are under way to replace jellium with a real gold surface, either flat or with a few gold atoms at the contact. We expect the latter will reduce the conductance substantially, in part because of the large path for tunneling.
The structure of the I-V curve can be understood in terms of the electronic structure of the molecule: The initial plateau in the current up to 0.7V is due to a low density of states between the right and left quasi-Fermi levels. The first peak is due to a resonance between the high-energy quasi-Fermi level and the molecular antibonding orbitals made up of pz orbitals, where z is the direction perpendicular to the plane of the ring. The valley in the conductance is due to the mismatch between the above antibonding states and the quasi-Fermi levels. This feature is a clear demonstration of resonant tunneling. The second peak at about 4V is again due to a resonant tunneling condition. The states involved in the present case are however of a different bonding nature.
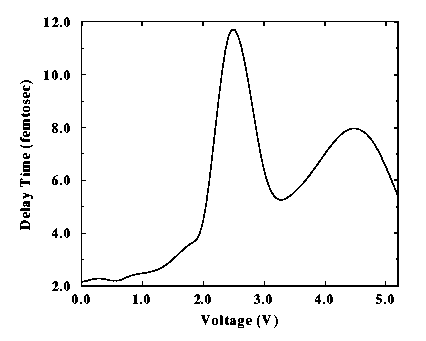
We also calculated the delay time of an electron tunneling through the above molecule in order to investigate the speed of operation of the device. The delay time as a function of the external voltage is plotted in Fig. 2.
| |
Fig. 2. Delay time of an electron tunneling through a benzene-1,4-dithiolate molecule as function of the external voltage. The peaks correspond to resonant tunneling conditions. |
|
|
The molecule allows electrons to tunnel within a few femtoseconds. The major delay appears at the bias where the first resonant tunneling occurs: at resonance the electrons spend most of their time going back and forth in the device before they escape into the external electrode. The delay times are generally much lower than the period of the first vibrational mode of the benzene ring, suggesting transport without scattering from vibrations. At resonance however the delay time is comparable to the C-H stretching vibrational mode, thus allowing a coupling of the electrons to the molecular vibrations. The overall effect would be to reduce and smooth the conductance for external voltages in the vicinity of the resonant tunneling condition. However, the speed of operation of the above device is still one to two orders of magnitude higher than conventional tunnel devices, thus showing promising applications in high-speed electronics.
The above calculations were done by means of a newly developed ab-initio approach [2]. The approach consists in calculating the Lippmann-Schwinger equation completely from first-principles, where the atoms are represented by ab-initio pseudopotentials and the electrodes by jellium layers. The procedure is self-consistent and allows calculating the current flowing through the device at any given external bias.
References
- M. Reed et al., Science 278, 252 (1997).
- N.D. Lang, Phys. Rev. B 52, 5335 (1995).
*Corresponding Address:
M. Di Ventra
Department of Physics and Astronomy, Vanderbilt University
P.O. Box 1807 - Station B, 6301 Stevenson Center, Nashville, TN 37235
phone: 615-343-1707; fax: 615-343-7697
e-mail: [email protected]
URL:
http://comped1.cas.vanderbilt.edu/~diventra/
|