Studies of Fullerenes and Carbon Nanotubes by an
Extended Bond Order Potential
by
Jianwei Che*, Tahir Çagin, William A. Goddard, III
Materials and Process Simulation Center, California Institute of Technology,
Pasadena, CA, 91125
This is a draft paper for the
Sixth Foresight Conference on Molecular Nanotechnology.
The final version was published in the special Conference issue of Nanotechnology:
Nanotechnology 10 (1999) 263-268.
Abstract
A novel method of implementing bond order potentials with long range non bond interactions has been developed. The extended bond order potential consistently takes bond terms and non bond terms into account. Not only it captures the advantages of the bond order potentials, (i.e. simulating bond forming and breaking), but also systematically includes the non bond contributions to energy and forces in studying the structure, and dynamics of covalently bonded systems such as graphite, diamond, nanotubes, fullerenes, hydrocarbons, their crystal and melt forms.
Non bond interactions is crucial to correctly predict the material properties.3 For instance, van der Waals forces are the main source for stabilizing the carbon nanotube bundles, fullerene crystals. Using this modified bond order potential, we studied the elastic properties and the plastic deformation processes of the single walled and double walled nanotubes. The bond order potential enables us to simulate a wide range of deformation of a nanotube under external loads.
The modified potential can also simulate molecular crystals and liquids, for instance, fullerene crystal and fullerene clusters. The interactions between fullerenes are non bond forces. Our modified potential consistently treats non bond and bonding interactions, which is applicable to a variety of problems. Moreover, the basic formulation is transferable to other bond order potentials and traditional valence force fields.
Introduction
In recent years, properties and structures of nano size materials have attracted many people's attention. Their unique properties and small dimensionality give very promising future for various potential applications. Since the discovery of carbon fullerenes1 and nanotubes2, large amount of theoretical and experimental work has been focused on characterization of these materials. Significant progress has been made toward understanding the properties and structures of nanometer scale materials. The typical size of these systems constitutes from dozens to millions of atoms. From theoretical calculation point of view, although the first principle calculation is feasible for small size systems, it is usually unpractical to carry it onto larger systems ( > 100 atoms). In this case, molecular dynamics simulation is one of the widely used theoretical methods to calculate various mechanical, thermodynamical, and even chemical properties of these systems.
The outcomes of molecular dynamics simulations are mainly determined by the interaction potentials (force fields) that are used in the calculations. It is crucial to know the validation regime of a certain potential function. In this paper, we will discuss our implementation for bond order potentials and its application to carbon based nano materials. Bond order potentials have the unique advantage over other traditional classical force fields (FF), since they are capable of describing the change of chemical bonds in a system. Therefore, bond order potentials offer an alternative way of investigating chemical reaction mechanisms, especially in condensed phases. Among various bond order potentials, Brenner bond order potential is parameterized specificly for carbon and hydrocarbon systems. It gives correct energetics not only for small gas phase molecules, but for condensed phase bulk systems (e.g, diamond crystal). Carbon nanotubes, fullerene molecules and clusters, and their crystals are of particular interests because of their unique structures and properties. Since these materials are all carbon based, it is straightforward to employ the Brenner bond order potential in MD simulations. Robertson et. al. have used this potential to calculate carbon nanotube energetics and compared the results with LDF calculations.4 In addition, it has been used for reactions between hydrocarbon species and diamond surfaces,5 chemical vapor deposition (CVD) of diamond films.6
In spite of its successes, the current form of bond order potentials, including Brenner force field, have limitations for accurate calculations of some material properties. One of the serious limitations is lacking of an uniform treatment for long range interactions, such as van der Waals and Coulomb forces. For a system without chemical reactions, long range interactions can contribute significantly to material property observables. For example, the existence of fullerenes and nanotubes is due to the inter molecular long range interactions. Consequently, the melting and crystallization temperatures, bulk densities, and thermal properties are wholly or partially affected by inter molecular interactions. Recently, self-assembled systems attract many people's interests. Long range forces also play a very important role in these system.
In this paper, we apply an extended Brenner bond-order dependent potential3 to study the properties of fullerenes and nanotubes. The implementation of long range interactions allow consistent treatment of all sorts of non-bond (NB) interactions. In addition, it retains all the advantages that bond order potentials have over traditional classical FF. Moreover, the implementation can also be applied to more traditional FFs.
The paper is organized as follows, in Sec. II, we briefly review the basic formulation of Brenner bond order potential and our implementation. The numerical results and discussions are given in Sec. III. Sec. IV concludes the paper.
Theory
The original Brenner bond order potential6 constitutes the summation of all pairwise interactions in a system, however, each pairwise interaction implicitly contains many body information from its immediate surroundings. The total potential energy is given by,
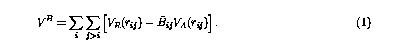
In the above equation, VR and VA are the repulsive and attractive part of the pairwise interaction. They have the general form of Morse potential,6
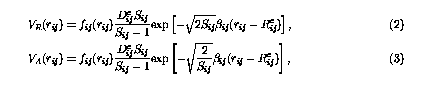
where
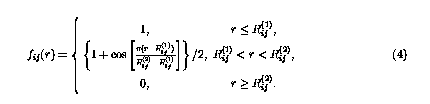
fij(r) is a cutoff function that smoothly goes to zero at 2.0 Å. It explicitly restricts the interaction within the nearest neighbors. 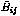 is the bond order parameter,6 is the bond order parameter,6
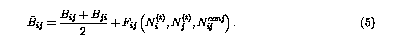
Bij and Fij are calculated from the coordination information of each pair ij. The values for all the parameters can be found in Brenner's original paper.6 It is clear that NB interactions are not included in the above equations. Several approaches has been considered to implement the original potential with NB forces. There are mainly two versions. One is the switching-on method, and the other is partition method. The former approach connects the short range covalent forces with long range NB interactions by a smooth function.7 The later approach partitions a system into different groups, and only imposes NB interactions among distinct groups.8 Since both approaches have certain limitations, we developed a new scheme to take NB forces into account.3
Using the new implementation, the total potential of a system is given by,
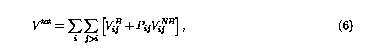
where Vij is simplified notation of Vij(rij), and Pij=Pji is a screening function that properly weights the NB contribution in the total energy. It is written as,3
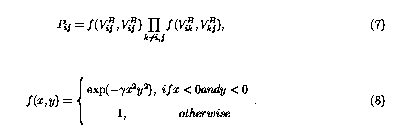
The superscript B denotes the covalent energy given by Brenner potential, and NB denotes the long range NB interactions. The force of each atom can be readily evaluated from Eq.(6),3

As we can see from the formulation, the source of NB interactions is not restricted. For instance, it can be van der Waals forces or Coulomb interaction, or both. Also the detailed functional form used in a specific system will not change the above fundamental equations. Therefore, our implementation offers a method with great flexibility. Moreover, it treats all the atoms in the system equivalently and consistently. In the next section, we will apply this method to a few carbon based nano structures.
Numerical Applications
As a limiting case, the implemented Brenner potential can also give the correct energetics for systems that NB interactions does not play a crucial role,3 such as an isolated small hydrocarbon molecule. In this case, it recovers the results of the original Brenner potential. However, since the original Brenner potential doesn't have NB interactions, it is not feasible to stabilize the crystal forms of molecular systems. Experimental results showed that the fullerene (C60) crystal is fcc form. Therefore, it is very important to take NB interactions into account. In this paper, we adopt the Leonard-Jones 12-6 potential for van der Waals interaction, and the parameters were obtained from the first principle calculation.9
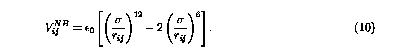
Using the implemented potential, the fcc crystal of C60 can be stabilized.The stabilization energy of the fcc crystal for C60 is 0.54 kcal/mol. Fig.1 shows the crystal unit cell obtained by the implemented Brenner potential. The cell parameter is 14.3 Å that is slightly larger than experimental value. The discrepancy is the result of inaccurate force constants for covalent bonds that give rise to a slightly large C60 molecule.
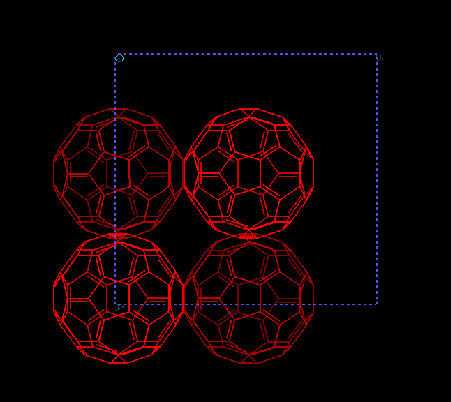 |
| Figure 1. A unit cell of the C60 crystal optimized by the extended bond order potential; the calculated lattice constant a = 14.4 Å. |
With the general implementation of nonbond interactions, various transport properties can also be readily calculated. Among the mechanical transport phenomena, thermal conductivity is one of the most difficult quantity to calculate, especially for solid state materials, because of the long phonon mean free path. However, under certain conditions, the thermal current correlation functions can be evaluated without extending the simulation system. Weak bonded solids and materials under high temperature have relatively short phonon mean free path that enables the numerical evaluation of thermal conductivity using small simulation cell. In addition to atomistic simulation, Boltzmann equation for transport phenonmena can also be used as a model to calculate material transport properties. Here, we will only demonstrate the ablility of the implemented potential to calculate thermal conductivity through atomistic simulation. The fundamental method is to make use of the Green-Kubo relation,
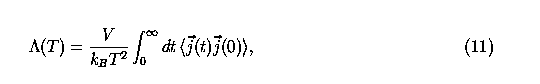
where j(t) is the thermal current,
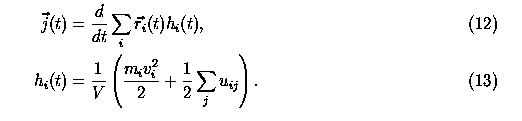
where hi is the site i energy density. In our simulation, it represents particle energy density. In general, thermal conductivity is a tensor. For an isotropic material, the average thermal conductivity is measured in experiments. At room temperature, the thermal conductivity of C60 crystal is 0.4 W m-1 K-1. Experiments also showed that the thermal conductivity of C60 crystal was nearly independent of temperature till 260 K. In Fig.2, we show the thermal current-current correlation function.
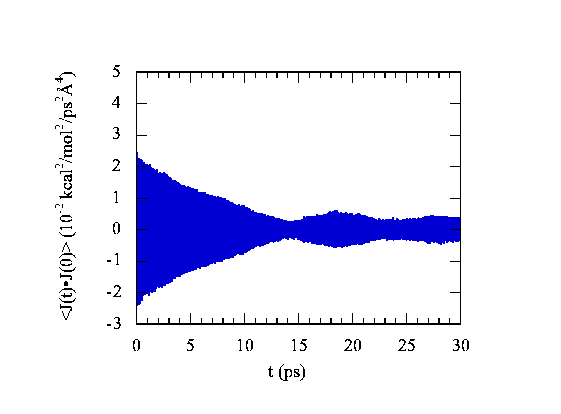 |
| Figure 2. The thermal current-current correlation function at room temperature for the C60 fcc crystal. |
The average thermal conductivity is obtained from the integration of the correlation function. The integration curve is depicted in Fig.3. The integration converges to a constant value with certain fluctuation due to the fluctuation of correlation function. The simulated result for the C60 thermal conductivity at room temperature is 0.4 W m-1 K-1 with the fluctuation of 20%.
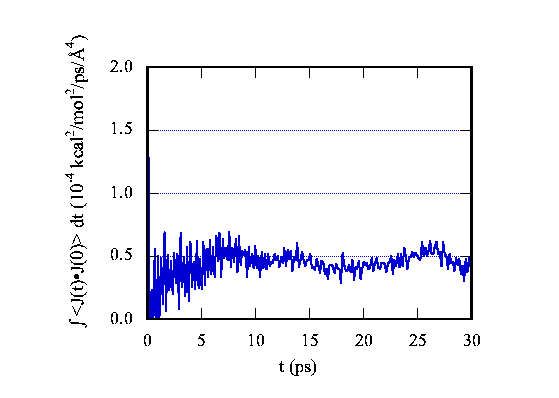 |
| Figure 3. The integration curve for the C60 crystal thermal current correlation function. The final value converges to 0.5 (differing from the thermal conductivity by a constant factor). |
Besides the crystal packing of nano structure, the accurate energetic calculation of a single walled nanotube (SWNT) also requires accurate NB interactions to be considered. Nanotube has been of great interest for many people since its discovery.10 Both theoretical predictions and experimental results indicate that nanotubes are one class of the strongest material known to date. The pure Brenner potential has been used for SWNT.4 However, since it has no NB interactions, it can not be applied to multi-walled nanotube (MWNT) directly. For MWNT, partition method can be employed to impose NB interactions only across different layers. The drawback of this method is preventing chemical reactions between atoms that belong to different layers. Even for SWNT, NB interactions can greatly affect the structures and energetics. Gao et. al.10 demonstrated that large SWNTs have collapsed form that is a result of intra-layer van der Waals interaction. Our implementation allows NB interactions to exist within each SWNT and across different tubes for MWNTs. Fig.4 shows the energetics of a (70,70) SWNT as a function of different conformations. For each structure, we applied constraints to restrict the distance between two atoms on the opposite sides across the diameter of the nanotube. Then each structure is optimized under the constraints. The energetics are depicted in Fig.4. The red line is obtained from implemented potential, and the blue line is calculated from the original Brenner potential. As we can see from the figure, the implemented potential gives the global optimal structure to be a collapsed form, and circular form is a metastable structure. The original bond order potential only predicts a single optimal structure that is circular form. The physical reason for the collapsed structure is that the van der Waals attraction between two flat area can lower the total energy. This flat region is an analog to graphite crystal stacking.
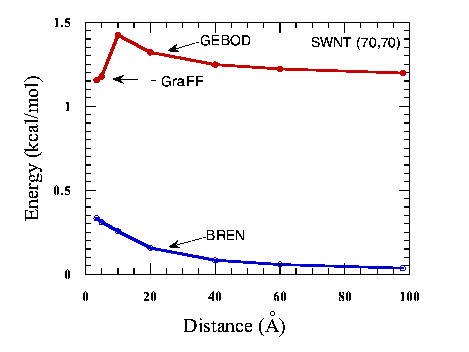 |
| Figure 4. The energetics of a single-walled nanotube (70,70) as a function of the tube conformation. The x-axis is the distance of constraints. |
For MWNTs, the inclusion of NB interactions is more important for many material properties. A double walled nanotube (DWNT) has an inter tube rotational mode. This mode is also a result of long range van der Waals interactions. Fig.5 plots the potential as a function of the relative rotational angle between two nanotubes. The outer tube is a (15,15) SWNT, and the inner one is a (10,10) tube. The space between them is 3.48 Å, which is close to the graphite inter layer distance (3.3545 Å). This motion is a very low frequency collective mode. When only original form of Brenner potential is used, the relative rotation resembles a completely free motion, since each tube is not able to feel the existence of another one. Only when are NB interactions considered, the correct dynamics can be extracted. From Fig.5, the rotational barrier is 10 cal/mol/layer. The potential has 5 repeating units, which is the maximum common divider of (10,10) tube and (15,15) tube repeating unit number.
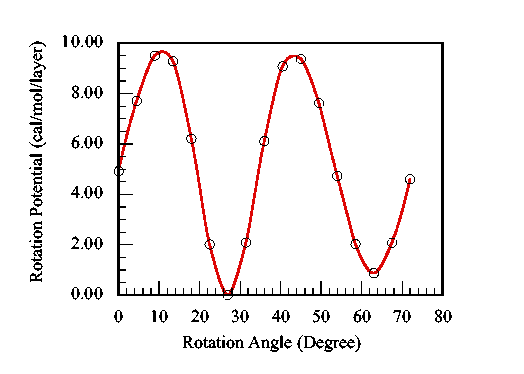 |
| Figure 5. The rotational potential as a function of relative rotational angle between inner and outer tubes The period for the rotational potential is 72°. |
One of the most important properties for nanotube is its high strength. Theoretical calculations and experimental results suggest that the Young's modulus for nanotube is on the order of 1TPa. As a comparison, the Young's modulus for diamond, the most known strong material, is about 1TPa. In our previous papers,2,3 we have reported the Young's modulus for SWNT and DWNT is around 940 GPa in the elastic regime. In future applications of nanotubes, people are interested the mechanical properties not only in the elastic regime, but in the plastic regime and the regime where mechanical failure occurs. For a SWNT, the uniaxial stretching mostly involves the covalent bond stretching and bending. In general, chemical bonds are orders of magnitude stronger than NB interactions, therefore, NB forces only contribute negligibly to the overall deformation. However, for MWNTs, condensed phase of SWNTs, and composite materials, NB interactions can be very important when the deformation involves the inter tube sliding, crossing, and compression. As an example in this paper, we only consider the mechanical behavior of a SWNT and a DWNT under dynamic stretching. For DWNT, no inter tube slipping is allowed. Therefore, NB interactions are not very crucial in the calculation.
A single nanotube of 20 layers long is used in the calculation. For a (10,10) tube, it contains 800 atoms, and (15,15)/(10,10) DWNT consists of 2000 atoms. One end of the tube is held stationary, and a constant velocity stretching (constant strain rate) is applied to the other end. Fig.6 shows the force and energy of the (10,10) SWNT during the stretching process. The red and blue curves are calculated from the implemented potential, and the black and green curves are obtained form the original Brenner potential. From the force evolution, it is clear that the tube undergoes elastic deformation first and then plastic deformation. The discontinuity occurs when the SWNT suffers the mechanical failure (disruption). As we can see from Fig.6, both the implemented and the original Brenner potential give very similar results. The implemented potential requires larger force and more energy because there is a small potential barrier created by NB interactions before disruption happens. However, the difference is small. More interestingly, Fig.6 shows that force constant starts to increase from 12ps as the system approaches the failure point. At a first glance, it is very similar to work-hardening. However, it is caused by the cutoff function fij in Eq.(2) and Eq.(3). The function artificially increases the force constant while it is turned on from Rij(1). Physically, the system should approach the failure point from plastic deformation regime without going through "work-hardening" process. This is another shortcoming in the Brenner potential. The cutoff function restricts the covalent interaction within 2 Å. Physically, it is not always the case. When a covalent bond is stretched, the two electrons in the bonding orbital are still spin-paired even two nuclei are separated by more than 2 Å. In other words, the covalent bonding still exists although it is weakened. In order to overcome this problem, a new scheme of calculating bonding information rather than a simple distance criterion is necessary.
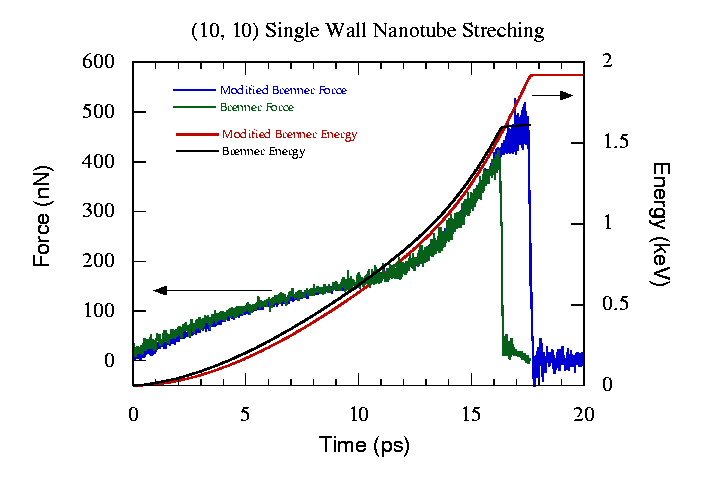 |
| Figure 6. The force and energy as a function of dynamic stretching time for (10,10) SWNT. |
The same dynamic stretching is also applied to a (15,15)/(10,10) DWNT. Since NB interactions are not important in this process, one can expect that the energy and force are simple addition of a (15,15) SWNT and a (10,10) SWNT. Fig.7 plots the energy and force as a function of time for the DWNT. Since the stretching is constant strain rate, the strain is proportional to its time scale. Because the stretching velocity is high, nanotubes do not have time to re-arrange themselves to form defect structures. From Fig.6 and Fig.7, the disruption occurs around 18ps. It is much shorter than the time scale to form defected structure that is on the order of nanosecond. Therefore, this type of dynamic stretching can be considered as an adiabatic process.
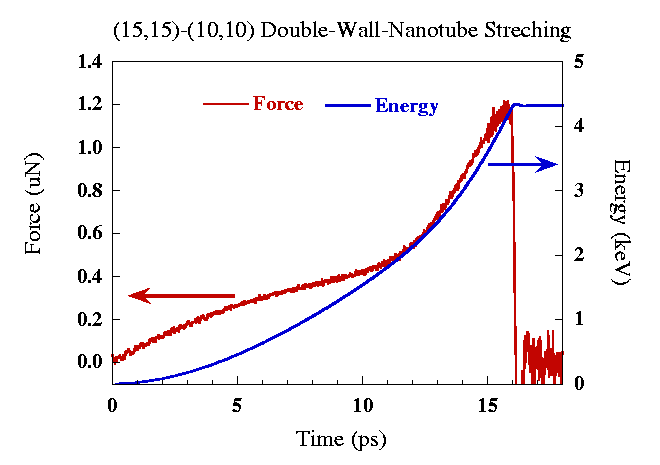 |
| Figure 7. The force and energy as a function of dynamic stretching time for (15,15)/(10,10) DWNT. |
From the figures, we can also see that the disruption occurs at very short time scale. Once the tube starts to break, the complete breakage happens extremely fast. Fig.8 and Fig.9 show the time evolution of the dynamic stretching processes.
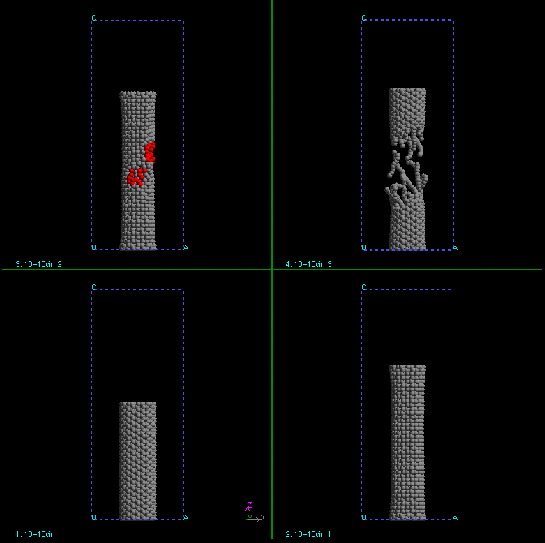 |
| Figure 8. The time evolution of dynamic stretching of (10,10) SWNT. |
|
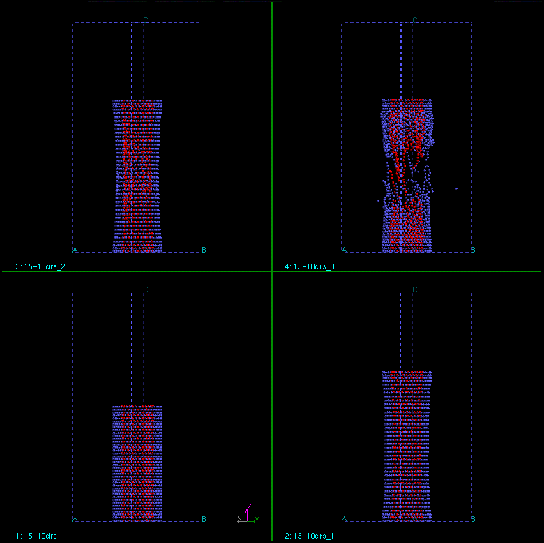 |
| Figure 9. The time evolution of dynamic stretching of (15,15)/(10,10) DWNT. The inner tube (10,10) has been colored red for clarity. |
Conclusions
In this paper, we applied an extended bond order potential to nanotube and fullerenes simulation. The extended potential considers NB interactions properly, and offers a generic scheme to implement bond order potential with long range interactions. Moreover, the modification of bond terms and NB terms can be separable. In other words, one can further improve bond potential or NB potential without worrying too much about the correlation effects. For example, we have discussed the problem of 2 Å cutoff in bond terms in the above section. To overcome this drawback, one might use other schemes which have much longer bonding range, the NB terms can remain untouched during the modification. Therefore, this is a robust scheme of include NB interactions. For the switching-on method, it needs to be reformulated or re-parameterized significantly if one alters bond terms. Using the new extended potential, we also discussed the conditions under which NB interactions are important and the situations where NB terms are not crucial. Through examining the bond order potential in more detail, we pointed out a few shortcomings of the existing potential and the directions that can be taken. We believe that bond order potentials are very powerful tools for classical simulations of material properties. A semiclassical bond order potential that has more quantum mechanical concepts will be the next generation of reactive potential.
Acknowledgments
This research was funded by a grant from NASA Computational Nanotechnology and by a grant from LLNL. The facilities of the MSC are also supported by grants from NSF (ASC 92-17368 and CHE 91-12279), ARO (MURI), ARO (DURIP), ONR (DURIP), Chevron Petroleum Technology Co., Asahi Chemical, Owens-Corning, Exxon, Chevron Chemical Co., Asahi Glass, Chevron Research Technology Co., Avery Dennison, BP America, and Beckman Institute.
References
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl, and R. E. Smalley, Nature, 318, 162 (1985).
- S. Iijima, ``Helical microtubules of graphitic carbon,'' Nature 354, 56 (1991).
- J. Che, T. Çagin, and W. A. Goddard III, ``Implementation of bond order potentials with nonbond interactions,'' Theoretical Chem. Acc. (1998), In press.
- D. H. Robertson, D. W. Brenner, and J. W. Mintmire, ``Energetics of nanoscale graphitic tubules,'' Phys. Rev. B 45, 12592 (1992).
- R. C. Mowrey, D. W. Brenner, B. I. Dunlap, J. W. Mintmire, and C. T. White, J. Phys. Chem. 95, 7138 (1991).
- D. W. Brenner, ``Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films,'' Phys. Rev. B 42, 9458 (1990).
- D. W. Brenner, D. H. Robertson, M. L. Elert, and C. T. White, ``Detonations at nanometer resolution using molecular dynamics,'' Phys. Rev. Lett. 70, 2174 (1993).
- D. W. Brenner, J. A. Harrison, C. T. White, and R. J. Colton, ``Molecular dynamics simulations of the nanometer-scale mechanical properties of compressed Buckminsterfullerene,'' Thin Solid Films 206, 220 (1991).
- Y. J. Guo, N. Karasawa, and W. A. Goddard III, ``Prediction of fullerene packing in C60 and C70 crystals,'' Nature 351, 464 (1991).
- G. Gao, T. Çagin, and W. A. Goddard III, Nanotechnology 9, 184 (1998).
|