Computational Studies of the Interaction of H/H2 with Diamond and Silicon Surfaces
by
Stephen P. Walch*, a, William A. Goddard IIIb, and Tahir Caginb
aELORET, 690 W. Fremont Ave, Suite 8, Sunnyvale, CA 94087-4202
bMaterials and Process Simulation Center, Beckman Institute (139-74), California Institute of Technology, Pasadena, CA 91125
*Corresponding author: [email protected]
This is a draft paper for the
Sixth
Foresight Conference on Molecular Nanotechnology.
The final version has been submitted
for publication in the special Conference issue of Nanotechnology.
Abstract.
Calculations have been carried out for the reaction of H and H2 with the diamond 100, 110, and 111 surfaces and the silicon 100 surface. Among reactions that were considered are symmetry constrained and non-symmetry constrained addition of H2 to a surface dimer, abstraction of an H atom from a surface C or Si atom by a gas phase H atom, and addition of an H atom to a surface dimer.
For the symmetry constrained addition of H2 to a surface dimer the barriers are 0.34 aJ (49 kcal/mol), 0.30 aJ (43 kcal/mol), and 0.10 aJ (14 kcal/mol) for diamond 100 and 110 and silicon 100, respectively.
The non-symmetry constrained addition of H2 to a surface dimer has only been fully characterized for the silicon 100 surface. Here the best estimate of the barrier with respect to the surface dimer plus H2 is 0.16 aJ (23.4 kcal/mol). This process involves partial conversion of the SiSi dimer to two isolated carbenes and addition of H2 to the carbene center. The barrier for this process is predicted to be much higher in the case of diamond 100 due to a larger dimerization energy (best estimate is > 0.49aJ (70 kcal/mol) ).
For the abstraction reaction written in the direction H2 + CCH --> H + HCCH, the barrier decreases and the reaction becomes increasingly exothermic in the order 100, 110, 111. These differences relate to the increase in CH bond strength with 111 > 110 > 100, and these differences in turn relate to the differences in overlap of the dangling bonds of a dimer pair.
It is found that the computed energetics for these reactions are strongly correlated with the overlap of the radical orbitals of a dimer pair in a GVB(pp) calculation.
I. Introduction.
A problem in nanotechnology is the need to tie off surface dangling bonds with hydrogen atoms. One approach to this problem involves the use of tools to add H atoms one at a time. While this approach is useful for demonstration of the concept and could be used to construct small pieces of nanomachinery or possibly an assembler, a more efficient method would be useful in many cases. We have considered the possibility of reacting a bare surface with gas phase H2/H as a means to passify the surface. In this study, we have considered the diamond100, 110, and 111 surfaces and the silicon100 surface.
Cluster models for the three surfaces of diamond are shown in Fig. 1. The unrelaxed 100 surface has carbene like surface carbon atoms; however, for the relaxed surface these dimerize to give rows of surface dimers and there is a considerable amount of p bonding between the radical orbitals of the dimer. The 110 surface has zig-zag rows of carbon atoms with a dangling bond on each carbon atom. These dangling bonds are hybridized away from each other and thus interact less strongly than for the 100 surface. Finally, the 111 surface has surface carbon atoms arranged in a triangular pattern and the surface dangling bonds are well separated from each other (second nearest neighbor distance) leading to almost no interaction between adjacent dangling bonds. These qualitative features may be quantified by computing the overlap of adjacent dangling bonds in a GVB(pp) calculation (Bobrowicz and Goddard, 1977). The overlaps are 0.462, 0.292, and 0.016 for the diamond 100, 110, and 111 surfaces, respectively, and 0.322 for the silicon 100 surface. (Note that these are with a 6-31G basis set and all the overlaps would be increased for a larger basis set, but these results clearly indicate the trends.) As discussed elsewhere (Walch and Merkle, 1998), the different overlaps between adjacent radical orbitals result in different reactivity for the various surfaces. For example, in the reaction with a carbene, the diamond 111 surface behaves like a radical, but the diamond 100 surface behaves like a p bond. Based on the GVB(pp) overlaps, the 110 surface is expected to be somewhere in between the 100 and 111 surfaces in reactivity.

Fig. 1. Cluster models for the 100, 110, and 111 surfaces of diamond.
Fig. 2 shows some reactions of H2/H with the reconstructed diamond (100) surface. Reaction (1) is a 2+2 cycloaddition and is expected to have a barrier. The size of the barrier will decrease as the amount of p bonding in the surface dimer decreases. However, this process is expected to have a significant barrier for the diamond 100 and 110 surfaces. Reaction (2a) is addition of H2 to a surface carbene atom. This process may have a low barrier; however, it requires activating the surface to create the reactive site. Reaction (2b) is one reaction which may activate the surface by creating a surface carbene. Reaction (3) is a hydrogen abstraction process. This process requires that the dimer have some biradical character. Reaction (4) is a reaction of atomic H with the surface. This process will have only a small barrier but requires that the H2 be dissociated (as e.g. by reaction with a hot tungsten filament).
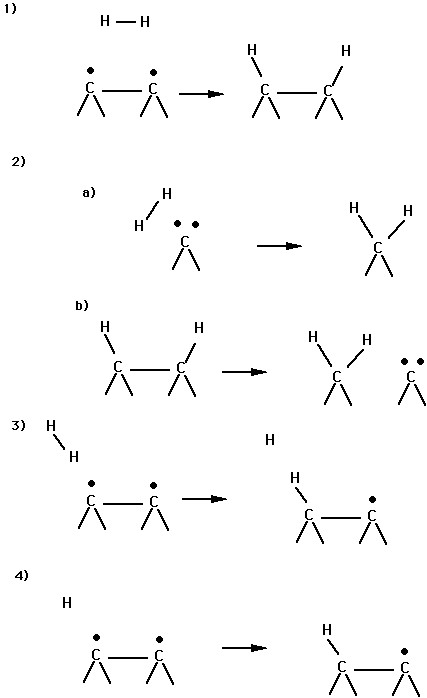
Fig. 2. Some reactions between H2/H and diamond (100) considered in this work.
A review of diamond surface chemistry is given in (Wei and Yates, 1995) and a discussion of H/silicon interactions is given in (Murty and Atwater, 1995).
II. Calculational Details.
The density functional theory (DFT) calculations used the B3LYP functional (Becke, 1993) and the 6-31G basis set (Ditchfield, Hehre, and Pople, 1971). the calculations were carried out with Gaussian94 (M. J. Frisch et al., 1995). In the case of Si, the Si1s22s22p6 core was replaced by an effective core potential (Stephens/Basch/Krauss ECP split valence-CEP-31G) (Stevens, Basch, and Krauss, 1984 ). The smallest basis set used here (denoted by small) is equivalent to the 6-31G basis set that was used for diamond. Because polarization functions are expected to be more important for silicon than for carbon, some larger basis sets were used for the silicon clusters. The medium basis set adds a set of d functions on silicon, and the large basis set also adds a set of p functions on H.
The calculations for ethylene + H2 were ab initio calculations. The stationary points were located with complete-active-space-self-consistent-field (CASSCF)/derivative methods (as implemented in SIRIUS/ABACUS) using the cc-pVDZ basis set (Dunning, 1989) and the energetics were determined with internally contracted CI (ICCI) (Werner and Knowles, 1988) (Knowles and Werner,1988) using the cc-pVTZ basis set (Dunning, 1989).
III. Discussion.
IIIa. Diamond 100 Surface.
a. Reaction (1).
Attempts to study reaction (1) using DFT were not initially successful. It is clear that reaction (1) requires a complete-active-space-self-consistent field (CASSCF) zero-order description since it involves a crossing of two configurations which differ by a double excitation. In order to gain insight into the problem, the gas phase reaction of ethylene with H2 was studied first using an ab initio approach. The starting point for these calculations was a CASSCF calculation with 4 electrons in 4 obitals; this was followed by an internally-contracted-configuration-interaction (ICCI) calculation with the cc-pVTZ basis set. The active orbitals correspond to the CC p bond in ethylene and the HH bond. Fig. 3 shows the results of this calculation. The process of adding H2 to ethylene in a C2v constrained geometry is seen to have a barrier in excess of 0.69 aJ (100 kcal/mol). This is a second order saddle point and allowing the symmetry to break leads to the other saddle point, which is basically an H abstraction process. (Note that the saddle point is above ethyl radical + H at the CASSCF level but ICCI places the saddle point below ethyl radical + H and this process probably occurs with no barrier.)
In order to model the analagous reaction with a dimer on the diamond (100) surface, a cluster was used which was a distorted ethylene with the CC distance fixed at the distance obtained for the singlet state in the DFT calculations. In one case the distorted ethylene was planar and in the other case the distorted ethylene had a tetrahedral configuration around the two carbons. The saddle points for H2 addition in a C2v constrained geometry were obtained from a CASSCF grid and the energetics were obtained from ICCI calculations as for the ethylene + H2. This gave barrier heights of 0.73 aJ (105.7 kcal/mol) for the planar cluster and 0.51 aJ(73.7 kcal/mol) for the tetrahedral cluster. As expected the tetrahedral cluster weakens the p bonding and lowers the barrier to addition. However, it is clear that this process will be unfavorable for a dimer on the diamond (100) surface. Table I shows energetics for the same process obtained with the DFT method. Here the distance above the surface was fixed and the surface dimer and H2 were allowed to relax at each distance in order to generate a C2v symmetry constrained minimum energy path. From Table I it is seen that DFT predicts a barrier of about 0.35 aJ (50 kcal/mol) for reaction (1).
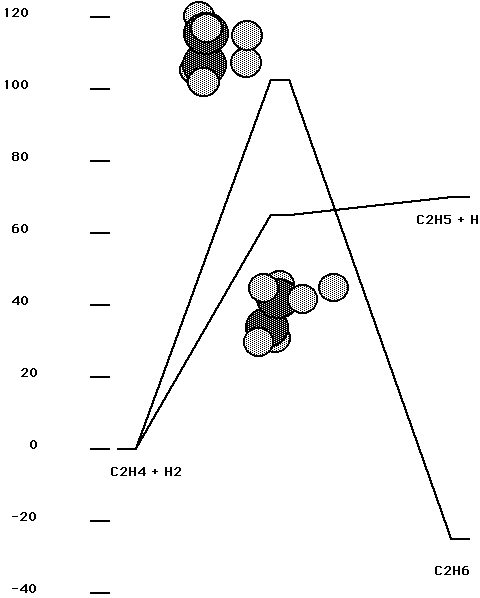
Fig. 3. Gas phase reaction of ethylene with H2. The vertical scale is energy in kcal/mol ( one kcal/mol = 0.00695 aJ). The same energy units are used in the remaining figures.
Table I. Diamond100 plus H2, C2v addition pathway (DFT calculations).
|
Ra |
DFT |
DE |
|
5.0 |
-351.08155 |
0.1 |
|
4.0 |
-351.08060 |
0.7 |
|
3.0 |
-351.05959 |
13.8 |
|
2.75 |
-351.03903 |
26.7 |
|
2.5 |
-351.00352 |
49.0 |
|
2.25 |
-351.17777 |
-60.3 |
|
2.0 |
-351.23220 |
-94.5 |
a R is the perpendicular distance from the second layer of the cluster. Column 2 gives the density functional theory (DFT) energy in atomic units (Hartree). Column 3 gives the relative energy in kcal/mol (one kcal/mol = 0.00695 aJ). The same conventions are used in the remaining tables.
b. Reaction (2).
Reaction (2) should have a low barrier since it is a carbene insertion process. As a first model a small cluster (See Fig. 4) was used which had one surface carbon and two second layer carbon atoms. (With the second layer carbon atoms fixed at bulk values and tied off with H atoms.) The barrier for reaction (2) was found to be 0.01 aJ (1.4 kcal/mol) and the exothermicity for this reaction is 0.79 aJ (113.3 kcal/mol). (See Table II.) Several other processes were also studied for this cluster. The inversion barrier for a H atom bonded to the carbene atom is less than 0.0007 aJ (0.1 kcal/mol). The HCC2 moiety is only 10 deg. from planar, which is reasonable by analogy to CH3, and consistent with the small computed barrier. The barrier to abstraction of an H atom from a surface CH2 by H atom was found to be to be 0.07 aJ (9.6 kcal/mol) and the heat of reaction is 0.03 aJ (4.4 kcal/mol) (endothermic). The final question was the barrier for adding H atom to a surface CH. No barrier was found for this ( doublet + doublet reaction) and the bond strength is found to be 0.73 aJ(105.4 kcal/mol).
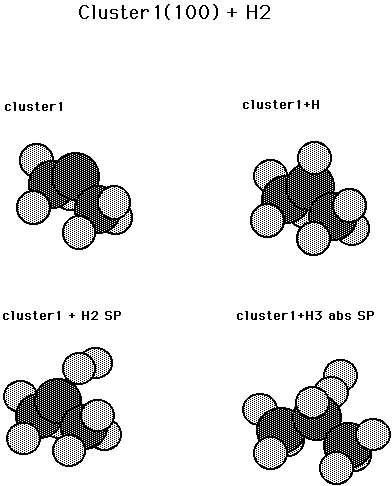
Fig. 4. Cluster model for a carbene site on diamond 100 plus H/H2. There is one surface C atom and two subsurface C atoms. Cluster1 is the bare cluster., cluster1+H is the monohydrogenated cluster, cluster1 + H2 SP is the saddle point for addition of H2 via a carbene process, and cluster1 + H3 abs sp is the saddle point for H atom abstraction from the dihydrogenated cluster.
Table II. Energetics for cluster1(100) + nH.
|
structure |
DFT |
DE |
|
cluster1 |
-117.75947 |
|
|
H |
-0.50027 |
|
| |
-118.25974 |
0.0 |
|
cluster1 + H |
-118.44733 |
-117.7 |
|
cluster1 + H SP |
-118.44732 |
-117.7 |
|
cluster1 |
-117.75947 |
|
|
H2 |
-1.17548 |
|
| |
-118.93495 |
0.0 |
|
cluster1+H2 sp |
-118.93265 |
1.4 |
|
cluster1 + H2 |
-119.11556 |
-113.3 |
|
cluster1 + H |
-118.44733 |
|
|
H2 |
-1.17548 |
|
| |
-119.62281 |
0.0 |
|
cluster1+H + H2 abs sp |
-119.60751 |
9.6 |
|
cluster1 + H2 |
-119.11556 |
|
|
H |
-0.50027 |
|
| |
-119.61583 |
4.4 |
|
cluster1 + H |
-118.44733 |
|
|
H |
-0.50027 |
|
| |
-118.94760 |
0.0 |
|
cluster1 + H2 |
-119.11556 |
-105.4 |
Fig. 5 and Table III show energetics for reaction (2b). (The cluster is shown in Fig. 1.) This process is assuming an entrance channel saddle point that leads to the structure labeled by min2 in Table III. This structure has two H atoms bonded to one C and the other carbon is a carbene site. An interesting feature of Fig. 5 is the low barrier for migration of a H onto a carbene center. This process is well known in gas phase chemistry (e.g. the isomerization of vinylidene to acetylene). (Walch and Taylor, 1995)
Table III. Energetics for reaction (2b).
|
structure |
DFT |
DE |
|
dimer singlet |
-349.90617 |
|
|
H2 |
-1.17548 |
|
| |
-351.08165 |
0.0 |
|
cluster100+h2.min2 |
-351.14608 |
-40.4 |
|
cluster100+h2.sp1 |
-351.14157 |
-37.6 |
|
cluster100+h2.min1 |
-351.24082 |
-99.9 |
a Note that cluster100 is shown in Fig. 1.
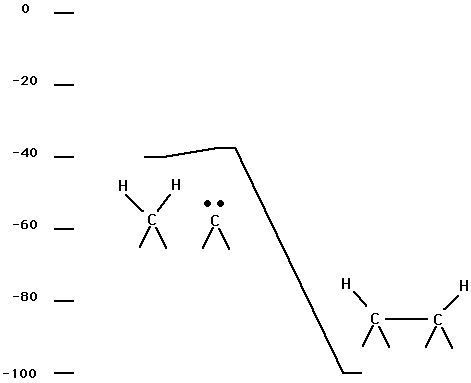
Fig. 5. Energetics for reaction (2b).
Another issue which was considered is the formation of a dihydride phase (i.e. two H atoms per surface C atom).
The structures which were considered here are shown in Fig. 7 and the energetics are shown in Table IVa. One question that hadn’t been considered before is the structure of the two minima for three and four H atoms on the 100 surface dimer. One speculation was that these structures would twist out-of-plane to minimize repulsions. However, calculations that were started with twisted geometries reverted to planar geometries when optimized. From the structure of the 4 H atom minimum it is clear that there would be significant interactions with an adjacent dimer site with 4 H atoms. Thus, for the extended surface with 2H per surface carbon there would be large non-bonded repulsions with the CH bonds of adjacent monohydrogenated dimers. The main effects of this non-bonded repulsion were included by placing CH2 groups in the locations of the nearer carbon of the adjacent surface dimers. (Here one CH bond is oriented along the CC bond of the adjacent dimer and the other CH bond is in the location of the CH bond of the adjacent surface dimer. See Fig. 6.) This is similar to what was done earlier in our studies of the monofluorinated silicon surface (Walch and Halicioglu, to be published). Table IVa gives computed energetics for the case without adjacent CH2 groups. Here it is seen that the binding energy for a third H atom to the mono hydrogenated dimer is 0.35 aJ (50.3 kcal/mol) or about half a CH bond strength. This is due to disruption of the CC p bond of the dimer. The barrier to H migration is only 0.10 aJ (13.7 kcal/mol), which is much less than the barrier of 0.44 aJ (63.8 kcal/mol) for H atom migration of a single H atom on a surface dimer. Adding another H atom to the dimer plus three H atoms leads to a CH bond strength of 0.68 aJ (98.4 kcal/mol) in the dimer plus 4 H atom cluster.
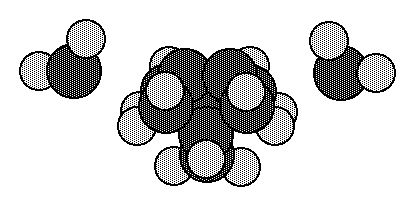
Fig. 6. Cluster for a surface dimer plus two adjacent CH2 groups to simulate the non-bonded repulsions with adjacent surface dimers.
A number of other results are also summarized in Table IVa. These include the barrier for carbene insertion of H2 into the structure (2b) of Fig. 2. This barrier is 0.04 aJ (6.3 kcal/mol), which is ~0.03 aJ (5 kcal/mol) larger than for reaction (2a).
From Table IVb it is seen that the presence of adjacent CH2 groups reduces the binding energies of H atoms to a surface dimer. The reductions are 0.01 aJ (2.0 kcal/mol), 0.09 aJ (12.4 kcal/mol), and 0.13 aJ (18.1 kcal/mol) for 2H, 3H, and 4H, respectively.
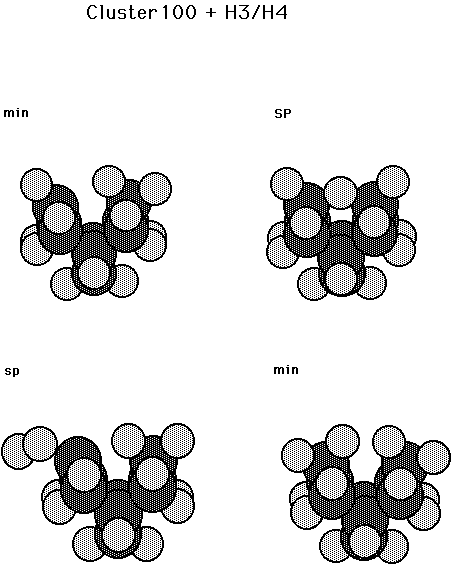
Fig. 7. Clusters for a surface dimer on diamond 100 plus three and four H atoms. The structure in the upper left is the minimum energy configuration for a dimer plus 3 H atoms. The structure in the upper right is the exchange saddle point for a dimer plus 3 H atoms. The structure in the lower left is the saddle point for H2 addition to an activated dihydrogenated surface dimer. The structure in the lower right is the minimum energy configuration for a surface dimer plus 4 H atoms.
Table IVa. Energetics for cluster100 + nH.
|
structure |
DFT |
DE |
|
cluster100 |
-349.90617 |
|
|
H |
-0.50027 |
|
| |
-350.40644 |
0.0 |
|
cluster100 + H |
-350.56623 |
-100.3 |
|
dimer singlet |
-349.90617 |
|
|
H2 |
-1.17548 |
|
| |
-351.08165 |
0.0 |
|
cluster100+H2.min2 |
-351.14608 |
-40.4 |
|
cluster100+H2.sp1 |
-351.14157 |
-37.6 |
|
cluster100+H2.min1 |
-351.24082 |
-99.9 |
|
cluster100 +H |
-350.56623 |
|
|
H |
-0.50027 |
|
| |
-351.06650 |
0.0 |
|
cluster100+H2.min1 |
-351.24082 |
-109.4 |
|
cluster100+H2.min1 |
-351.24082 |
|
|
H |
-0.50027 |
|
| |
-351.74109 |
0.0 |
|
cluster100+H3, exc sp |
-351.79945 |
-36.6 |
|
cluster100+H3 |
-351.82120 |
-50.3 |
|
cluster100+H2.min1 |
-351.24082 |
|
|
H2 |
-1.17548 |
|
| |
-352.32156 |
0.0 |
|
cluster 100+H4 sp |
-352.31146 |
6.3 |
|
cluster100+H4 |
-352.47834 |
-98.4 |
|
cluster100+H3 |
-351.82120 |
|
|
H |
-0.50027 |
|
| |
-352.32147 |
0.0 |
|
cluster100+H4 |
-352.47834 |
-98.4 |
Table IVb.
|
structure |
DFT |
DE |
|
cluster100+2ch2 |
-428.12925 |
|
|
H2 |
-1.17548 |
|
| |
-429.30473 |
0.0 |
|
cluster100+2ch2 + H2 |
-429.46072 |
-97.9 |
|
cluster100+2ch2 + H2 |
-429.46072 |
|
|
H |
-0.50027 |
|
| |
-429.96099 |
0.0 |
|
cluster100+2ch2 + H3
SP |
-430.01735 |
-35.4 |
|
cluster100+2ch2 + H3 |
-430.02139 |
-37.9 |
|
cluster100+2ch2 + H3 |
-430.02139 |
|
|
H |
-0.50027 |
|
| |
-430.52166 |
0.0 |
|
cluster100+2ch2 + H4 |
-430.64957 |
-80.3 |
c. Reaction (3).
Table V and Fig. 8 show the reaction of H2 with a surface dimer along an abstraction pathway. From Fig. 8 it is seen that the abstraction reaction is nearly thermoneutral and has a small barrier. The calculations were carried out for the triplet spin state. Fig. 8 also shows a singlet pathway. This pathway is constructed assuming that the barrier is comparable on the singlet surface. Note that for the reverse reaction either singlet or triplet spin is possible, and the saddle point region on the singlet surface is assumed to be an open shell singlet. Here it is seen that on the singlet surface, the barrier including the endothermicity is about 0.10 aJ (15 kcal/mol).
Table V. Energetics for abstraction of a H from cluster100+h.
|
structure |
DFT |
DE |
|
cluster100+h |
-350.56623 |
|
|
H |
-0.50027 |
|
| |
-351.06650 |
0.0 |
|
cluster100+h2.abs |
-351.05537 |
7.0 |
|
dimer triplet |
-349.88903 |
|
|
H2 |
-1.17548 |
|
| |
-351.06451 |
1.2 |
|
dimer singlet |
-349.90617 |
|
|
H2 |
-1.17548 |
|
| |
-351.08165 |
-9.0 |
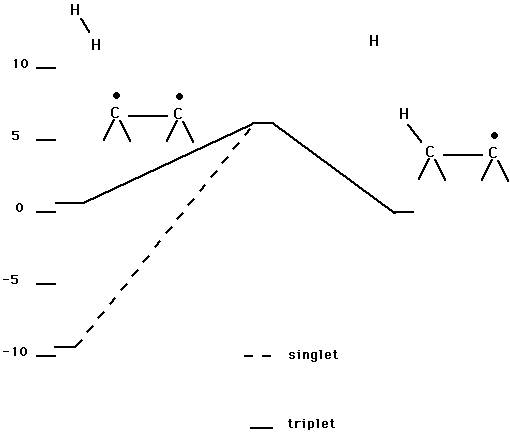
Fig. 8. Abstraction pathway for H2 reacting with a surface dimer on diamond (100). The singlet and triplet pathways are indicated.
Table VI and Fig. 9 show the process of adding a second H atom to the surface dimer via the abstraction pathway. This process occurs only on a doublet surface and has a small (about 0.04 aJ (6 kcal/mol) ) barrier.
Table VI. Energetics for abstraction of a H from min1.
|
structure |
DFT |
DE |
|
cluster100+h2.min1 |
-351.24082 |
|
|
H |
-0.50027 |
|
| |
-351.74109 |
0.0 |
|
cluster100+h3.sp1 |
-351.73166 |
5.9 |
|
cluster100+h |
-350.56623 |
|
|
H2 |
-1.17548 |
|
| |
-351.74171 |
-0.4 |
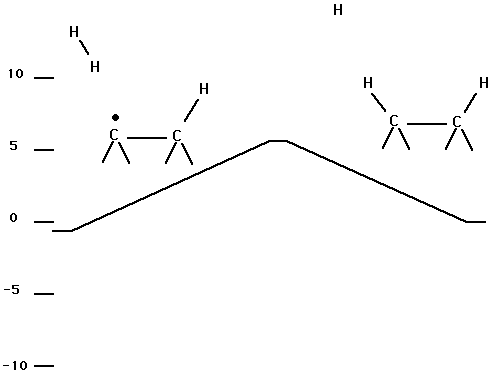
Fig. 9. Energetics for adding a second H atom to a dimer on diamond (100) via an abstraction process.
d. Reaction (4).
Table VII and Fig. 10 show energetics for adding an H atom to a dimer on diamond (100). DFT calculations show no barrier to addition of H to the singlet state of the dimer. Addition of a second H atom should also be a barrierless process. The exchange of an H atom between the two C atoms is also shown. This process has a barrier of 0.44 aJ (63.8 kcal/mol).
Table VII. Energetics for H atom plus dimer on diamond 100.
|
structure |
DFT |
DE |
|
cluster100 |
-349.90617 |
|
|
H |
-0.50027 |
|
| |
-350.40644 |
0.0 |
|
cluster100 +h |
-350.56623 |
-100.3 |
|
exchange sp |
-350.46461 |
-36.5 |
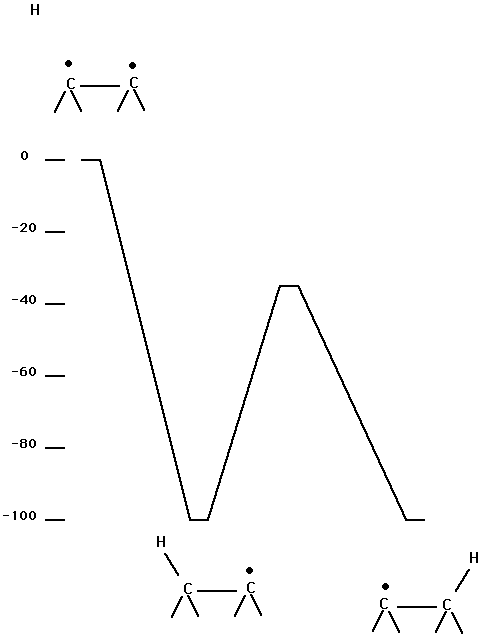
Fig. 10. Energetics for adding an H atom to a dimer on diamond (100).
IIIb. Diamond 110 Surface.
We now consider reactions on the diamond 110 surface. Fig. 11 shows the clusters that were used for this surface. This cluster has 4 surface and 6 second layer C atoms with the dangling bonds tied of with H atoms. Here the position of the 4 surface atoms was partially optimized (with the constraint that the three CC bond lengths remain the same). ( Calculations in which the CC bond lengths were allowed to be different indicated a slight energetic preference for an alternating structure.) The center two surface atoms of this cluster will be considered as a dimer pair. The computed energetics are given in Table VIII. From Table VIII it is seen that the CH bond strength for adding one H atom to the dimer pair (cluster110b+h3) is 0.76 aJ (109.6 kcal/mol) and the exchange barrier is 0.50 aJ (72.0 kcal/mol) as compared to 0.44 aJ (63.8 kcal/mol) for the 100 surface. This larger barrier probably results from more hydridization of the radical orbitals away from each other as compared to the 100 surface. The overlap of adjacent singlet coupled radical orbitals is 0.292 in a GVB(pp) calculation and the singlet to triplet excitation energy is 0.05 aJ (7.3 kcal/mol) for the diamond110 surface, compared to 0.462 and 0.22 aJ (32 kcal/mol) for the diamond100 surface. The abstraction barriers were also computed for abstracting an H atom from the mono and dihydrogenated dimer (cluster110b+h2+h2.abs and cluster110b+h3+h2.abs, respectively). The abstraction barriers are 0.04 aJ (5.2 kcal/mol) and 0.05 aJ (6.8 kcal/mol) and the heats of reaction are 0.0007 aJ (0.1 kcal/mol) and 0.01 aJ (1.9 kcal/mol) for the mono and dihydrogenated dimer, respectively.
Calculations were also carried out for a symmetry constrained addition of H2 across the dimer. These results are given in Table IX. Here it is seen that the barrier for the symmetry constrained approach is ~0.30 aJ (43 kcal/mol). For diamond 100 the barrier was ~0.34 aJ (49 kcal/mol). This is consistent with the smaller overlap of the p bond for the 110 surface compared to the 100 surface.
Table IX. Energetics for cluster110b+h2 + H2. (C2v)
|
structure |
R |
DFT |
DE |
|
cluster110b+h2 |
|
-390.47045 |
|
|
H2 |
|
-1.17548 |
|
| |
|
-391.64593 |
0.0 |
| |
3.5 |
-391.62828 |
11.1 |
| |
3.25 |
-391.61083 |
22.0 |
| |
3.0 |
-391.57688 |
43.3 |
| |
2.75 |
-391.66756 |
13.6 |
| |
2.5 |
-391.73221 |
-54.1 |
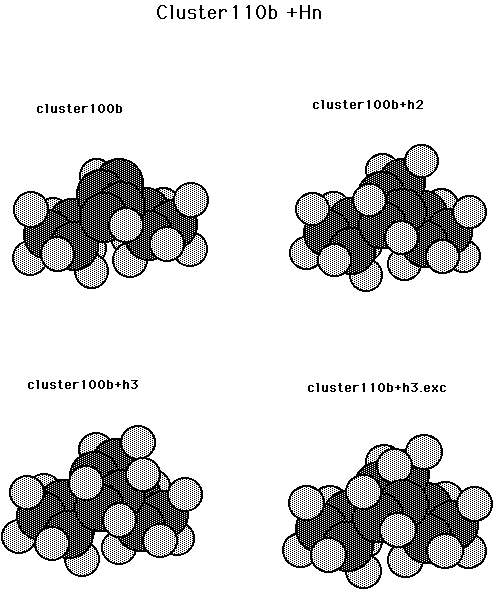
Fig. 11. Cluster model for the diamond 110 surface plus 1-4 H atoms.
Table VIII. Energetics for cluster110b + nH.
|
structure |
DFT |
DE |
|
cluster110b |
-389.15909 |
|
|
H |
-0.50027 |
|
| |
-389.65936 |
0.0 |
|
cluster110b+h2 |
-390.47045 |
|
|
H |
-0.50027 |
|
| |
-390.97072 |
0.0 |
|
cluster110b+h3 |
-391.14543 |
-109.6 |
|
cluster110b+h3,exc. |
-391.03057 |
-37.6 |
|
cluster110b+h3 |
-391.14543 |
|
|
H |
-0.50027 |
|
| |
-391.64570 |
0.0 |
|
cluster110b+h4 |
-391.81757 |
-107.9 |
|
cluster110b+h2 |
-390.47045 |
|
|
H2 |
-1.17548 |
|
| |
-391.64593 |
0.0 |
|
cluster110b+h3.abs |
-391.63764 |
5.2 |
|
cluster110b+h3 |
-391.14543 |
|
|
H |
-0.50027 |
|
| |
-391.64570 |
0.1 |
|
cluster110b+h3 |
-391.14543 |
|
|
H2 |
-1.17548 |
|
| |
-392.32091 |
0.0 |
|
cluster110b+h4 abs |
-392.31004 |
6.8 |
|
cluster110b+h4 |
-391.81757 |
|
|
H |
-0.50027 |
|
| |
-392.31784 |
1.9 |
Table IX. Energetics for cluster110b+h2 + H2. (C2v)
|
structure |
R |
DFT |
DE |
|
cluster110b+h2 |
|
-390.47045 |
|
|
H2 |
|
-1.17548 |
|
| |
|
-391.64593 |
0.0 |
| |
3.5 |
-391.62828 |
11.1 |
| |
3.25 |
-391.61083 |
22.0 |
| |
3.0 |
-391.57688 |
43.3 |
| |
2.75 |
-391.66756 |
13.6 |
| |
2.5 |
-391.73221 |
-54.1 |
IIIc. Diamond 111 Surface.
Calculations were also carried out for several processes on the diamond 111 surface. The cluster used here has two surface carbons and one second layer carbon. (See Fig. 12 and Table X) The two surface dangling bonds are separated by second nearest neighbor distances and interact only very weakly. This weak interaction is evident in the first and second CH bond energies which are 0.81 aJ (116.1 kcal/mol) and 0.81 aJ (115.9 kcal/mol), respectively. Also these binding energies are significantly larger than on the 100 and 110 surfaces, which also is expected from the free radical character of the surface dangling bond on the 111 surface, compared to the other surfaces, where there is some p bonding. The barrier for abstraction is calculated to be 0.03 aJ (4.0 kcal/mol) and the reaction is exothermic by 0.04 aJ (6.1 kcal/mol). The exothermic nature of the abstraction reaction is due to the greater CH bond strength for the 111 surface.
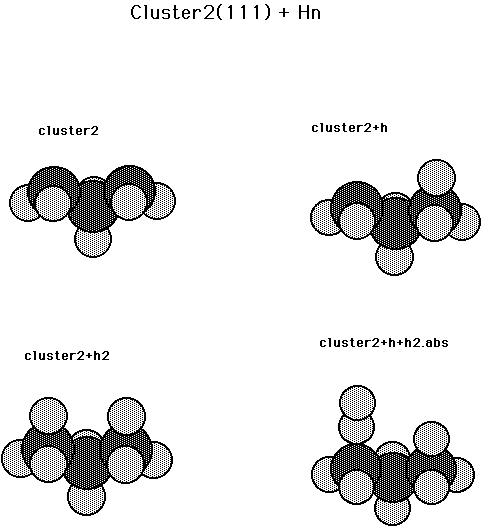
Fig. 12. Cluster model for diamond 111 + Hn.
Table X. Energetics for cluster2(111)+h2 + H2.
|
structure |
DFT |
DE |
|
cluster2 |
-117.74505 |
|
|
H |
-0.50027 |
|
| |
-118.24532 |
0.0 |
|
cluster2+h |
-118.43039 |
-116.1 |
|
cluster2+h |
-118.43039 |
|
|
H |
-0.50027 |
|
| |
-118.93066 |
0.0 |
|
cluster2+h2 |
-119.11536 |
-115.9 |
|
cluster2+h |
-118.43039 |
|
|
H2 |
-1.17548 |
|
| |
-119.60587 |
0.0 |
|
cluster2+h+h2, abs |
-119.59943 |
4.0 |
|
cluster2+h2 |
-119.11536 |
|
|
H |
-0.50027 |
|
| |
-119.61563 |
-6.1 |
IIId. Silicon 100 Surface.
Fig. 13 and Table XI show results for a dimer on the reconstructed Si100 surface plus one to four H atoms. The binding energies shown in Fig. 13 are for the processes: Si100+H --> Si100H, Si100+H2--> HSiSiH, HSiSiH+H--> HSiSiH2, and HSiSiH+H2--> SiH2SiH2, respectively. Table XI also gives the individual bond strengths for HSiSiH. Here it is seen that the bond strengths for the first and second SiH bonds are 0.60 aJ (86.1 kcal/mol) and 0.58 aJ (83.4 kcal/mol), respectively. Note that these are De’s (i.e they do not include an estimate of vibrational zero-point effects). An estimate of these effects may be taken from earlier work ( Wu and Carter, 1991), which leads to D0’s of 0.54 aJ (78.1 kcal/mol) and 0.57 aJ (81.4 kcal/mol) for the first and second SiH bond strengths. This suggests a significant preference for adding a second H atom to the same dimer. From Fig. 13 it is also seen that the DE’s for Si100+H2--> HSiSiH and HSiSiH+H2--> SiH2SiH2 are 0.40 aJ (57.8 kcal/mol) and 0.10 aJ (15.0 kcal/mol), respectively. This compares to 0.69 aJ (99.9 kcal/mol) and 0.68 aJ (98.4 kcal/mol) for the analagous structures on the diamond 100 surface. Thus, the formation of a dihydride phase is less likely on silicon as compared to diamond.
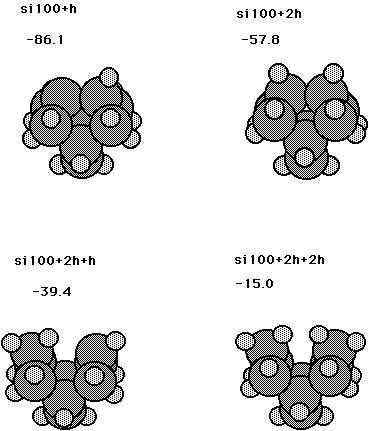
Fig. 13. Structures for a dimer on the silicon 100 surface plus one to four H atoms. The numbers are binding energies in kcal/mol for the processes Si100+H --> Si100H, Si100+H2--> HSiSiH, HSiSiH+H--> HSiSiH2, and HSiSiH+H2--> SiH2SiH2, respectively
Table XI. Si100+nH (medium basis)
|
Si100 (singlet) |
-42.00983 |
|
|
H |
-0.50027 |
|
| |
-42.51010 |
0.0 |
|
Si100 + H |
-42.64727 |
-86.1 |
|
Si100 + H |
-42.64727 |
|
|
H |
-0.50027 |
|
| |
-43.14754 |
0.0 |
|
Si100+H2 |
-43.28041 |
-83.4 |
|
Si100 (singlet) |
-42.00983 |
|
|
H2 |
-1.17854 |
|
| |
-43.18837 |
0.0 |
|
Si100+H2 |
-43.28041 |
-57.8 |
|
Si100+H2 |
-43.28041 |
|
|
H |
-0.50027 |
|
| |
-43.78068 |
0.0 |
|
Si100+2H+H |
-43.84346 |
-39.4 |
|
Si100+H2 |
-43.28041 |
|
|
H2 |
-1.17854 |
|
|
Si100+H2+H2 |
-44.45895 |
0.0 |
|
Si100+2H+2H |
-44.48278 |
-15.0 |
Table XII and Fig. 14 show the energetics for adding H2 to a dimer on the silicon (100) surface. This is a C2v constrained minimum energy pathway obtained in the same way as for the results for diamond 100 + H2 shown in Table I. Here it is seen that the barrier is only about 0.10 aJ (14 kcal/mol) (using the small basis set) as compared to 0.35 aJ (50 kcal/mol) for diamond. Table XIII and Fig. 14 show energetics for the abstraction process analagous to reaction (3) for the diamond 100 surface. Here it is seen that abstraction of one H from H2, to give a single H bonded to the surface dimer plus H atom, is uphill by 0.15 aJ (22 kcal/mol) (using the small basis set), but there is no barrier other than the endothermicity. Thus, for Si100 it is more favorable to add an H2 via the C2v constrained approach leading to the Si100 + H2 structure than to transfer a single H to the surface (abstraction pathway). ( See Fig. 14.) Table XIII and Fig. 15 show a non C2v constrained addition pathway. Here it is seen that the addition barrier is 0.16 (23.5 kcal/mol) (using the small basis set) and subsequent steps are all below the reactants energy. The structures for min1, sp1, min2, and sp3 are given in Fig. 16. Details of the pathways are given in Fig. 17 for sp3, which is the non C2v addition pathway, and in Fig. 18 for sp1, which is the second barrier shown in Fig. 15.
The non-symmetry constrained addition of H2 to the surface dimer (reaction (2)) differs for diamond and silicon. In the diamond case, this reaction can happen in two ways. One occurs by activation of the surface dimer by moving both H atoms onto one dimer carbon atom (reaction (2b) ). (CHCH --> CH2C) This process is endothermic by 0.41 aJ (59.5 kcal/mol) and requires 0.43 aJ (62.3 kcal/mol) to surmount the barrier. The resulting CH2C structure is 0.28aJ (40.4 kcal/mol) below the surface dimer plus H2. The resulting localized carbene center can add an H2 by a carbene insertion process with a low barrier ( about 0.04 aJ (6 kcal/mol) ) leading to a dihydride structure. The other mechanism involves a competition between the dimerization energy per dimer and the low barrier to carbene insertion into H2 for a localized carbene site. However the dimerization energy for diamond 100 is computed to be 0.48 aJ (69.3 kcal/mol) per dimer and therefore the barrier to addition would be a predicted to be greater than 0.49 aJ (70 kcal/mol). (This saddle point has not been located.) In the silicon case, all the saddle point structures for this reaction have been located. The first step is addition of H2 to a surface dimer leading to a SiH2Si structure. In the silicon case the dimerization energy per surface dimer is 0.12 aJ (17.4 kcal/mol) which is consistent with the barrier for non-symmetry constrained addition of H2 which is 0.16 aJ (23.4 kcal/mol) with the largest basis set. Thus, for the silicon case symmetry constrained and non-symmetry constrained addition of H2 have comparable barriers. In the silicon case the non-symmetry constrained addition is followed by rearrangement to a SiHSiH structure, which is analagous to reaction (2b) in the diamond case.
Table XIIIb gives energetics with the large basis set for the processes dicussed above. Here it is seen that the best estimates of the barrier heights are 0.17 aJ (24.7 kcal/mol) for abstraction, 0.13 aJ (19.4 kcal/mol) for symmetry constrained addition, and 0.13 aJ (23.4 kcal/mol) for non-symmetry constrained addition. This leads to a bottom-of-well to bottom-of-well estimate of 0.58 aJ (82.9 kcal/mol) for the barrier to desorption of H2. (via the non-symmetry-constrained pathway). The experimental estimates of this quantity are 0.31 aJ (45 kcal/mol) to 0.46 aJ (66 kcal/mol). However, the computed quantity does not include vibrational zero-point effects or the destabilization of the HSiSiH structure by interaction with adjacent monohydrogenated dimers. Both of these effects would be expected to significantly reduce the computed activation energy for desorption of H2. Therefore, we can not say how well the present results agree with the experimental activation energy until estimates of these quantities are obtained. An important result in the present work is that the non-symmetry-constrained and symmetry-constrained pathways have very similar barrier heights. Thus, both pathways may be occuring simultaneously.
Table XIV and Fig. 19 show energetics for a single H atom bonded to a surface dimer and for the exchange of this atom between the two si atoms of the dimer. Here it is seen that the most reliable estimate of the barrier to exchange (large basis set results) is 0.30 aJ (43.6 kcal/mol) for silicon 100 as compared to 0.44 aJ (63.8 kcal/mol) for diamond 100.
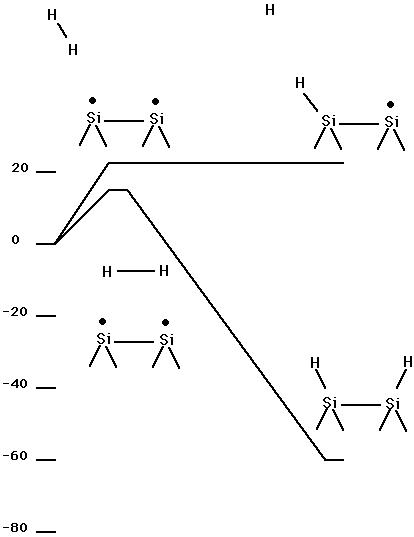
Fig. 14. Energetics for Si100 + H2 (symmetry constrained approach and abstraction pathway).
Table XII. Si100 plus H2, C2v addition pathway (small basis).
|
Ra |
DFT |
DE |
|
5.0 |
-42.98212 |
0.9 |
|
4.0 |
-42.96978 |
8.7 |
|
3.75 |
-42.96341 |
12.7 |
|
3.5 |
-42.96195 |
13.6 |
|
3.25 |
-42.96182 |
13.6 |
|
3.0 |
-43.06702 |
-52.4 |
a R is the perpendicular distance from the second layer of the cluster.
Table XIIIa. Si100 plus H2 (small basis).
|
Si100 (singlet) |
-41.80809 |
|
|
H2 |
-1.17548 |
|
| |
-42.98357 |
0.0 |
|
Si100 + H2 abstr. sp |
-42.94918 |
21.6 |
|
Si100 + H min. |
-42.44862 |
|
|
H |
-0.50027 |
|
| |
-42.94862 |
21.9 |
|
Si100 + H2 min1 |
-43.07823 |
-59.4 |
|
Si100 + H2 sp1 |
-43.01594 |
-20.3 |
|
Si100 + H2 min2 |
-43.03474 |
-32.1 |
|
Si100 + H2 sp3 |
-42.94617 |
23.5 |
Table XIIIb. Si100 plus H2 (large basis).
|
Si100 (singlet) |
-42.02589 |
|
|
H2 |
-1.17854 |
|
| |
-43.20443 |
0.0 |
|
Si100 + H2 abstr. sp |
-43.16502 |
24.7 |
|
Si100 + H min. |
|
|
|
H |
|
|
|
Si100 + H2, C2v , 3.75 |
-43.17375 |
19.3 |
|
Si100 + H2, C2v , 3.625 |
-43.17358 |
19.4 |
|
Si100 + H2, C2v , 3.5 |
-43.17351 |
19.4 |
|
Si100 + H2, C2v , 3.25 |
-43.17500 |
18.4 |
|
Si100 + H2, C2v , 3.0 |
-43.28242 |
-48.9 |
|
Si100 + H2 min1 |
-43.29922 |
-59.5 |
|
Si100 + H2 sp1 |
-43.23628 |
-20.0 |
|
Si100 + H2 min2 |
-43.25019 |
-28.7 |
|
Si100 + H2 sp3 |
-43.16714 |
23.4 |
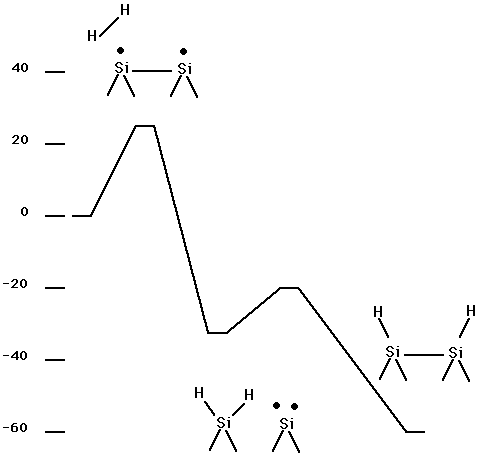
Fig. 15. Energetics for Si100 + H2( non C2v approach).
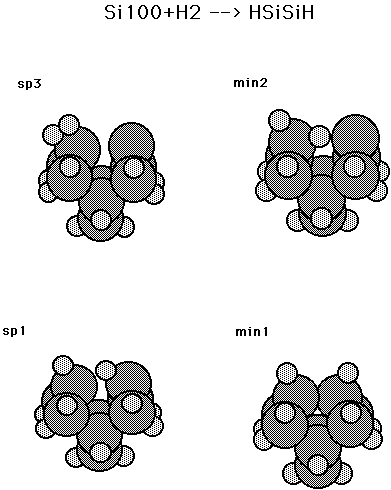
Fig. 16. Stationary point structures for Si100 + H2 (non C2v approach).
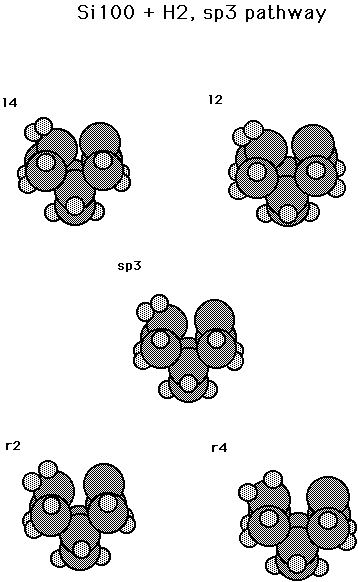
Fig. 17. Minimum energy path for the reaction Si100 + H2 --> sp3 --> min2.
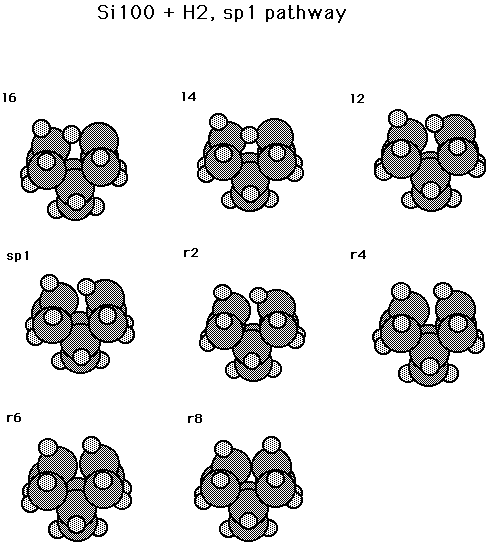
Fig. 18. Minimum energy path for the reaction min2 --> sp1 --> min1.
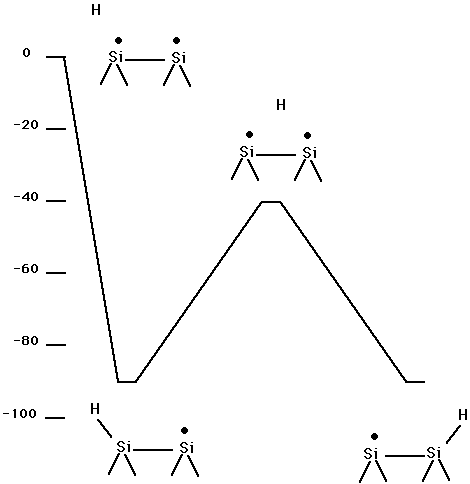
Fig. 19. Energetics for Si100 + H.
Table XIVa. Si100 +H (small basis).
|
Si100 (singlet) |
-41.80809 |
|
|
H |
-0.50027 |
|
| |
-42.30836 |
0.0 |
|
Si100 + H exchange |
-42.37274 |
-40.4 |
|
Si100 + H min. |
-42.44862 |
-88.0 |
Table XIVb. Si100 +H (large basis).
|
Si100 (singlet) |
-42.02589 |
|
|
H |
-0.50027 |
|
| |
-42.52616 |
0.0 |
|
Si100 + H exchange |
-42.59533 |
-43.4 |
|
Si100 + H min. |
-42.66474 |
-87.0 |
IV. Conclusions.
We have looked at the reaction of H2 with the diamond 100, 110, and 111 and silicon 100 surfaces using a cluster model and DFT with the B3LYP functional. For each of the four surfaces we have studied the symmetry constrained and non-symmetry constrained addition of H2, the addition of H atom, and abstraction of an H atom from a surface CH by a gas phase H atom.
For the dimer reconstructed 100 surfaces we considered the reactions shown in Fig. 2. The symmetry constrained addition of H2 to the surface dimer (reaction (1)) is found to have barriers of 0.35 aJ (50 kcal/mol) and 0.10 aJ (14 kcal) for diamond and silicon 100 surfaces, respectively. (Using comparable double-zeta like basis sets.) For the silicon surface the barrier increases to 0.13 aJ (19.4 kcal/mol) with addition of polarization functions to both Si and H.
The non-symmetry constrained addition of H2 to the surface dimer (reaction (2)) differs for diamond and silicon. In the diamond case, one possibility is activation of the surface dimer by moving both H atoms onto one dimer carbon atom (reaction (2b) ). (CHCH --> CH2C) This process is endothermic by 0.41 aJ (59.5 kcal/mol) and requires 0.43 aJ (62.3 kcal/mol) to surmount the barrier. The resulting CH2C structure is 0.28 aJ (40.4 kcal/mol) below the surface dimer plus H2 and there is a small barrier ( about 0.04 aJ (6 kcal/mol) ) to addition of H2 to the localized carbene center to give a dihydride structure. The other possibility involves a competition between the dimerization energy per surface dimer and the small barrier for addition of H2 to a localized carbene center. However the dimerization energy for diamond 100 is computed to be 0.48 aJ (69.3 kcal/mol) per dimer and therefore the barrier to addition would be predicted to be greater than 0.49 aJ (70 kcal/mol). (This saddle point has not been located.) In the silicon case, all the saddle point structures for this reaction have been located. The first step is non-symmetry constrained addition of H2 to a surface dimer leading to a SiH2Si structure. Here the dimerization energy per surface dimer is 0.12 aJ (17.4 kcal/mol). This is consistent with the barrier for non-symmetry constrained addition of H2 which is 0.16 aJ (23.4 kcal/mo)l with the largest basis set. Thus, for the silicon case, symmetry constrained and non-symmetry constrained addition of H2 have comparable barriers. The non-symmetry constrained addition is followed by rearrangement to a SiHSiH structure.
For hydrogen abstraction by a gas phase H atom (reaction(3) ), there is a barrier of about 0.04 aJ (6 kcal/mol) with respect to H atom and the surface CHC species. For the process H2 + CC --> H + HCC the reaction is approximately thermoneutral on the triplet surface, but is uphill by about 0.07 aJ (10 kcal/mol) on the singlet surface in addition to the 0.04 aJ (6 kcal/mol) barrier. For the silicon triplet surface, this reaction is uphill by 0.15 aJ (22 kcal/mol), but there is no barrier other than the endothermicity. For the reaction H2 + CCH --> H + HCCH, the reaction is exothermic by 0.003 aJ (0.4 kcal/mol) and has an approximately 0.04 aJ (6 kcal/mol) barrier.
For the addition of a H atom to a surface dimer, there is no barrier for either the diamond or silicon surfaces.
Another process which was studied for the 100 surface is the barrier to exchange of an H atom ( HCC --> CCH or HSiSi --> SiSiH). The barriers here are 0.44 aJ (63.8 kcal/mol) and 0.30 aJ (43.6 kcal/mol) for diamond and silicon, respectively.
Important differences are seen between diamond and silicon with respect to the abstraction process. For the diamond surface, the abstraction process has the lowest barrier (other than the H atom addition, which is barrierless on both surfaces) but the C2v constrained addition of H2 has a large barrier. On the silicon surface, this ordering is reversed and the symmetry constrained addition of H2 has a lower barrier than the abstraction reaction. These differences are related to the properties of the surface dimer, especially the lower amount of p bonding for the silicon surface as compared to the diamond surface.
Calculations were also carried out for the analagous reactions on diamond 110 and 111 surfaces. For the diamond 110 surface the exchange barrier is larger 0.50 aJ (72.0 kcal/mol) than the 0.44 aJ (63.8 kcal/mol) barrier obtained for the 100 surface. The barrier to symmetry constrained addition of H2 is 0.30 aJ (43 kcal/mol) as compared to 0.34 aJ (49 kcal/mol) for the 100 surface. Both of these results are related to the lower overlap of the two orbitals of the dimer pair p bond for the 110 surface as compared to the 100 surface. Abstraction barriers were also computed. These are 0.04 aJ (5.2 kcal/mol) and 0.04 aJ (5.8 kcal/mol) for the reactions H + CHC --> H2 + CC and H + CHCH --> H2 + CCH, respectively, and the reactions are approximately thermoneutral. For the 110 surface the non-symmetry constrained addition of H2 would be expected to be unfavorable, since breaking the dimer bond involves breaking at least 3 CC bonds.
For the 111 surface of diamond, the surface dangling bonds are only very weakly interacting, and the only reaction that was considered likely on this surface is abstraction of a H atom by a gas phase H atom. If the reaction is written in the direction H2 + CCH --> H + HCCH the barrier is 4.0 kcal/mol and the reaction is exothemic by 0.04 aJ (6.1 kcal/mol). Note that for the diamond 100 surface the analogous reaction is endothermic by 0.07 aJ (10 kcal/mol) in addition to the 0.04 aJ (6 kcal/mol) barrier (on the singlet surface), while for the 110 surface there is a 0.3-0.4 aJ (5-6 kcal/mol) barrier and the reaction is essentially thermoneutral, and for the 111 surface the barrier is reduced to 0.03 aJ (4 kcal/mol) and the reaction is exothermic. These differences relate to the increase in CH bond strength with 111 > 110 > 100, and these differences in turn relate to the differences in overlap of the dangling bonds of a dimer pair.
References.
- A. D. Becke, J. Chem. Phys., 98, 5648 (1993).
- F. W. Bobrowicz and W. A. Goddard III, in Methods of Electronic Structure Theory, Modern Theoretical Chemistry, Plenum, New York, 1977, pp 79-126.
- R. Ditchfield, W. J. Hehre, and J. A. Pople, J. Chem. Phys., 54, 724(1971).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople, Gaussian 94, Revision D.1, Gaussian, Inc., Pittsburgh PA, 1995.
- T. H. Dunning, Jr., J. Chem. Phys., 90, 1007 (1989).
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 145, 514(1988).
- M. V. Ramana Murty and H. A. Atwater, Phys. Rev. B, 51, 4889(1995).
- SIRIUS is an MCSCF program written by H. J. Jensen and H. Agren and ABACUS is an MCSCF derivatives program written by T. Helgaker, H. J. Jensen, P. Jørenson, J. Olsen, and P. R. Taylor.
- W. Stevens, H. Basch, and J. Krauss, J. Chem. Phys., 81, 6026(1984).
- S. P. Walch and R. C. Merkle, Nanotechnology, 1998.
- J. Wei and J. T. Yates, Jr., Critical reviews in Surface Chemistry, 5, 1 (1995).
- H. -J. Werner and P. J. Knowles, J. Chem. Phys. , 89, 5803(1988).
- C. T. Wu and E. A. Carter, Chem. Phys. Lett, 185, 172 (1991).
|