Control and Placement of Molecules via Self-Assembly
by
P. A. Lewis, Z. J. Donhauser, B. A. Mantooth,
R. K. Smith, L. A. Bumm, K. F. Kelly, P. S. Weiss*
Department of Chemistry, The Pennsylvania State University
University Park, PA 16802-6300
*E-mail address for corresponding author: [email protected]
Web page for corresponding author: http://stm1.chem.psu.edu/
This is a draft paper for the
Eighth
Foresight Conference on Molecular Nanotechnology.
The final version has been submitted
for publication in the special Conference issue of Nanotechnology.
Abstract
We use self- and directed assembly to pattern organic monolayers on the nanometer scale. The ability of the scanning tunneling microscope to obtain both nanometer-scale structural and electronic information is used to characterize patterning techniques, to elucidate the intermolecular interactions that drive them, and to probe the structures formed. We illustrate three successful approaches: (1) phase separation of self-assembled monolayers by terminal and internal functionalization, (2) phase separation of self-assembled monolayers induced by post-adsorption processing, and (3) control of molecular placement by insertion into a self-assembled monolayer.These methods demonstrate the possibilities of patterning films by exploiting the intrinsic properties of the molecules.We employ these methods to prepare matrix-isolated samples to probe molecular electronic properties of single and bundled molecules.
Introduction
Control and stabilization of molecular assemblies at the nanometer scale are crucial steps in the fabrication of molecular-scale devices. Current techniques such as photolithography or electron beam lithography (Rai-Choudhury, 1997) and 'soft lithography' (Xia and Whitesides, 1998; Zhao et al., 1997) are limited in their resolution and cannot reproducibly achieve patterns with dimensions at the nanometer scale. On the other end of the spectrum, single-molecule manipulation has been successfully demonstrated using scanning probe microscopy, but is unable to produce devices in parallel and is still too time-consuming to be practical as a fabrication technique (Becker et al., 1987; Eigler and Schweizer, 1990; Weiss and Eigler, 1993; Gimzewski and Joachim, 1999; Hla et al., 2000). We anticipate the need to combine the speed and versatility of lithographic techniques with the resolution of single-molecule manipulation in order to construct commercially viable molecular devices.
We and others have developed and utilized methods that exploit the inherent chemical, physical, and thermodynamic properties of molecules for facile means to pattern surfaces at the molecular level using self-assembly techniques. Self-assembly is a natural phenomenon that can be observed in many biological, chemical, and physical processes (Ulman, 1991; Ulman, 1996). This method has been recently explored as a way to produce supramolecular assemblies in a straightforward manner and has been strategically manipulated to form nano-scale patterns. (Allara, 1995; Bain et al., 1989; Bain and Whitesides, 1989b; Ulman, 1991; Ulman, 1996) The most commonly studied and well-characterized systems are alkanethiolate self-assembled monolayers (SAMs) on Au{111} (Dubois and Nuzzo, 1992; Poirier, 1997). Alkanethiolate SAMs form spontaneously on Au{111} through chemisorption of the S head group to the Au surface. The monolayers interact on the surface through van der Waals forces that occur amongst adjacent alkyl chains. The origin of the stability of SAMs is thus two-fold, due to the covalent S-Au bond and the attractive van der Waals forces between the methylene groups. As a result of the intrinsic stability of these systems, SAMs are known to have a low defect density and resist degradation in air.
The process of self-assembly lends itself naturally to controlling the local placement of molecules. In particular, SAMs have been used as model systems for fabricating structures with controlled geometries (Hong et al., 1999; Liu et al., 2000; Zhao et al., 1997), as well as essential components in the actual device structure (Collet, 2000; Collett, 1998; Boulas, 1996). Multi-component SAMs formed by codeposition of two or more adsorbates from solution have been investigated for their patterning potential (Bain and Whitesides, 1988; Bain and Whitesides, 1989a; Bain and Whitesides, 1989b; Folkers et al., 1994; Smith et al., in press; Stranick et al., 1996; Stranick et al., 1994). When more than one adsorbate is considered, it is necessary to account for the interactions between the different adsorbates and whether these will favor mixing or separation.
Whitesides and co-workers studied multi-component SAMs codeposited from solution using contact angle measurements and X-ray photoelectron spectroscopy (XPS) to examine possible phase separation (Folkers et al., 1994). It was concluded from this study that SAMs adsorbed from a solution at equilibrium containing a long-chain and a short-chain alkanethiolate will not separate into discrete domains but rather randomly intermix on the surface. Other studies conducted by Bain and Whitesides using SAMs coadsorbed from alkanethiolates possessing different terminal functionality produced similar conclusions (Bain and Whitesides, 1988; Bain and Whitesides, 1989b).
However, these analyses involved analytical methods that are ensemble techniques (e.g., XPS, infrared spectroscopy, ellipsometry, and wettability measurements) which determine averaged, 'macroscopic' properties of the sample. Insofar as molecular level patterning is concerned, where the size regime for structures at the nanometer scale is desired, it is necessary to use techniques that are able to distinguish molecular level characteristics. With the development of scanning probe microscopies, such as scanning tunneling microscopy (STM) (Binnig et al., 1982) and atomic force microscopy (AFM) (Binnig et al., 1986), it is possible to gain local information at the atomic-scale and obtain real-space images of surfaces. This has proven to be enormously beneficial in studies of patterning such as phase separation of SAMs, where previous analytical methods were able to measure only ensemble averages.
We are concerned with the design and control of systems such as these, and have been successful in demonstrating various methods for patterning SAMs using self-assembly to form the structures and STM (along with ensemble-type techniques) to determine the success of the method. This paper outlines recent research conducted by our group involving patterning of SAMs, namely phase separation due to terminal and internal functionality, phase separation induced by post-adsorption processing, and controlled placement of individual molecules by insertion.
Phase Separation Driven by Terminal Functionality

larger picture |
| Figure 1. Two sequential STM images taken 37 min apart showing a SAM containing islands of CH3(CH2)15S- (topographically lower, shown as darker) surrounded by CH3OCO(CH2)15S- (topographically higher, shown as brighter) areas.The SAM was formed from a solution of 75% CH3OCO(CH2)15SH/25% CH3(CH2)15SH on Au{111}. Domain coalescence is observed at this time scale (indicated by the two arrows). The reverse process (one large domain separating into two smaller ones) was not observed. Imaging conditions: Vtip=-1 V, I=1 nA, 500 Å x 310 Å. |
Stranick et al. conducted an extensive study of the phase separation of multi-component SAMs (Stranick et al., 1994). The components in the SAMs were -S(CH2)15CH3 and-S(CH2)15OCOCH3, which are similar in length and chemical activity and differ only in their terminal functional groups. STM was used to obtain real-space images of the phase-separated domains. Although it was possible to characterize the average film composition using ensemble techniques (e.g., XPS, IR spectroscopy, and ellipsometry), the imaging capability of a scanning probe method was useful in determining whether the system phase separated on the nanometer scale. Figure 1 is an STM image of phase-separated domains of these two components in the SAM. The topographically higher areas (shown as brighter) were assigned as the methyl ester terminated alkanethiolate domains whereas the lower areas are the hexadecanethiolate domains. The STM topographic height difference between the areas corresponds to ~1 Å with the methyl ester terminated molecules appearing higher. In this instance, the domains were small (nanometers) such that in previous ensemble-averaged measurements, the films would have appeared to be randomly mixed. However, the STM images indicate that phase separation is occurring with no external modification necessary.
To understand phase separation, the enthalpic and entropic contributions must be considered. Since entropy favors mixing when two or more components are involved, the formation of discrete domains indicates that the enthalpic contributions of the system must outweigh the entropic contributions. Also, it is commonly thought that adsorption from solution is reversible, with exchange processes between the molecules on the surface and those in solution continuously occurring (Folkers et al., 1994). Therefore, as the system approaches equilibrium, the formation of domains indicates that this is a lower energy state as compared to random intermixing.
In addition to this, the prospect of surface diffusion must also be considered. In this study, coalescence of hexadecanethiolate domains was shown to occur over a period of less than 1 hr (Figure 1). Additionally, the divergence of one large domain into two smaller ones was not observed. This indicates that there is some degree of lateral motion on the surface. Although this diffusion is slow with respect to adsorption from the solution, it can be hypothesized from these observations that the lateral motion is working toward formation of segregated domains. Thus, we assume that both exchange processes in solution and lateral surface diffusion after removal from solution are important in SAMs that form phase separated domains.
As the domains were found to coalesce slowly and reduce their curvature over time (Stranick, 1994), we conclude that the system had not reached equilibrium. Keeping these films from reaching equilibrium by controlling the rates of motion and exchange has since become a cornerstone in our strategy for controlling nanometer-scale structure (Bumm, 1999a).
Although the tail groups in these molecules are only weakly interacting, this driving force is enough to induce separation at the molecular level. This contradicts earlier studies of multi-component SAMs in which it was inferred from averaging measurement techniques that random mixing of the two components in a SAM accurately represented the surface composition. Studies involving other mixtures of molecules differing only in the end group (i.e., -CH3, -CO2CH3, -OH, and -CN) were also performed (Stranick et al., 1996). These films also separated into different domains to some degree, with the exception of the -OH and -CN terminated systems. Since the basis for separation in these systems is the interaction between end groups, it follows that the degree of end group polarity will drive the separation process. It is thus possible to control the phase separation of molecules by selecting end groups with large differences in polarity.
Phase Separation Driven by Internal Functionality
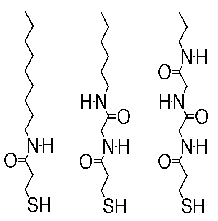
larger picture |
| Figure 2. Schematic of the family of molecules used in the amide-alkanethiolate studies. Left to right: 1ATC9, 2ATC6, and 3ATC3. Note the capability to form two hydrogen bonds per amide group with appropriately aligned adjacent molecules. |
Phase separation can also be influenced by the internal functionalization of the molecules in a multi-component SAM. Recently, we have conducted research on amide-containing alkanethiolate SAMs (e.g., 3-mercapto-N-nonylpropionamide, or 1ATC9) codeposited with a non-functionalized alkanethiolate (e.g., decanethiolate) (Smith et al., in press). A schematic of the family of the amide-containing molecules is shown in Figure 2. Because of the internal amide functional group, adjacent molecules can form stabilizing hydrogen bonds between the carbonyl and amino moieties that will influence their interactions on the surface.
Hutchison and co-workers have demonstrated the formation of stable monolayers of amide-containing alkanethiolates using XPS, contact angle goniometry, IR spectroscopy, and electrochemical methods (Clegg and Hutchison, 1996; Clegg and Hutchison, 1999; Clegg et al., 1999). It was concluded that hydrogen bonding of the amide functional groups and van der Waals interactions of the alkyl chains play an equally important role in determining whether the SAMs will be well-ordered. In monolayers of amide-containing alkanethiolates with alkyl chains containing fewer than 15 methylene units, the SAMs were disordered; however, it was shown that the underlying amide layer still appeared well-ordered.
These systems were subsequently studied using STM to gain an understanding of the ordering of the monolayers at the nanometer scale (Smith et al., in press). Upon codeposition with decanethiol, phase separation occurred spontaneously at room temperature. Figure 3 is a STM image of a film coadsorbed from a 1:1 solution of 1ATC9 and decanethiolate on Au{111}. The 1ATC9 molecules are physically higher than decanethiolate molecules by ~3.3 Å when adsorbed on the surface. These regions show up in the STM images as topographically higher (brighter areas). Both the 1ATC9 and decanethiolate domains are highly ordered and adopt ( 3x3)R30° lattice structures. Phase separation was observed over varying molar ratios of 1ATC9 and decanethiol solution (1% / 99%, 5% / 95%, 25% / 75%, 50% / 50%, 75% / 25%, 95% / 5%, 99% / 1% 1ATC9/decanethiol) (Lewis et al., in preparation). In films adsorbed from solutions containing a lower concentration of 1ATC9 molecules (i.e., 1%, 5%, 25% 1ATC9), the 1ATC9 domains appear to be forming at domain boundaries and substrate defects since these nucleation sites are areas in the film that are accessible to the 1ATC9 molecules. Additionally, the films are stable in air. Thus the placement and number of molecules adsorbed on the surface can be controlled by monitoring the defect density as well as the concentration of adsorption solution. 3x3)R30° lattice structures. Phase separation was observed over varying molar ratios of 1ATC9 and decanethiol solution (1% / 99%, 5% / 95%, 25% / 75%, 50% / 50%, 75% / 25%, 95% / 5%, 99% / 1% 1ATC9/decanethiol) (Lewis et al., in preparation). In films adsorbed from solutions containing a lower concentration of 1ATC9 molecules (i.e., 1%, 5%, 25% 1ATC9), the 1ATC9 domains appear to be forming at domain boundaries and substrate defects since these nucleation sites are areas in the film that are accessible to the 1ATC9 molecules. Additionally, the films are stable in air. Thus the placement and number of molecules adsorbed on the surface can be controlled by monitoring the defect density as well as the concentration of adsorption solution.
 |
 |
| Figure 3. (A) 1000 Å x 1000 Å STM image of a phase-separated SAM coadsorbed from a 1:1 solution of 1ATC9 (topographically higher areas) and decanethiol (topographically lower areas). An enlarged view (250 Å x 250 Å) of the boxed area in (A) is shown in (B). Imaging conditions: Vtip=-1 V, I=1 pA. |

larger picture |
| Figure 4. 800 Å x 800 Å STM image demonstrating phase-separation within a SAM coadsorbed from a 1:1 solution of 2ATC6 (topographically higher areas) and decanethiol (topographically lower areas).Imaging conditions: Vtip=-1 V, I=3 pA. |
Unlike the studies performed by Hutchison and co-workers, the monolayers in these films are well-ordered independent of the length of the alkyl chain (9 methylene units). However, it is important to note that both the substrates and the analytical methods used in each study were different and are likely responsible for the apparent discrepancy between the two studies. The substrates used for the IR studies performed by Hutchison and co-workers were Au/Cr/SiO2 as compared to the more ordered Au-on-mica for the STM studies. The disorder seen in the IR samples with fewer than 15 methylene units may be a result of the polycrystalline Au/Cr/SiO2 substrate and not of the inherent ordering of the system.
Hydrogen bonding is a logical explanation for the observed phase separation since these interactions would be much more favorable than interactions between the polar amide group and nonpolar alkyl chains of the decanethiolate molecules that would occur in random mixing of the two species. Also, van der Waals interactions of the alkyl chain overlayer in the 1ATC9 molecules provides additional stabilization to the molecules. In SAMs coadsorbed from solutions containing 3-mercapto-N-(N'-n-hexylacetamido)propionamide (referred to as 2ATC6) and decanethiol, phase separation was also noted(Figure 4) (Lewis, in preparation).However, STM measurements of mixed SAMs of 3-mercapto-N-(N'-(N''-n-propylacetamido)acetamido)propionamide (referred to as 3ATC3) and decanethiol showed that monolayers were not ordered, despite the 3ATC3 molecules having six opportunities for hydrogen bonding with their nearest 3ATC3 neighbors (two per amide functional group). As the number of methylene units in the alkanethiolates decreases, the SAMs require longer adsorption times to form ordered monolayers because of the absence of stabilizing van der Waals forces (Dubois and Nuzzo, 1992). Therefore, it follows that the flexible alkyl chains play an integral role in ordering the SAM; well-ordered films (and thus phase-separated domains) will not form without van der Waals contributions as well as hydrogen bonds between adjacent amide-alkanethiolate molecules.
These mixed SAMs prepared from amide-containing alkanethiolates and n-alkanethiolates show potential for patterning, as in a platform for adsorbing other molecules. Although the SAMs possess the same exposed functionality at the surface of the film, the differing underlying layers lead to different desorption characteristics, sticking coefficients, and domain stability. Because the alkanethiolate domains are held on the surface less strongly, they will desorb more readily than the amide-containing molecular domains. Thus, patterning can be achieved this way, with the ability to control the desorption of part of the monolayer in order to replace it with another component onto the surface (Clegg et al., in preparation).
Phase Separation due to Post-Adsorption Processing

larger picture |
Figure 5. 250Å x 250Å STM image of a SAM adsorbed on Au{111} from a solution of 95% CH3(CH2)9SH/ 5% CH3(CH2)11SH. The topographically higher areas correspond to the dodecanethiolate molecules and are randomly interspersed throughout the decanethiol matrix. Imaging conditions: Vtip=+1 V, I=10 pA. |
Along with spontaneous separation of SAMs into molecular domains, post-adsorption processing of monolayers can also be used in directing the assembly process. Bumm et al. focused on thermal annealing of a (dodecanethiolate) SAM followed by adsorption of a different alkanethiol (decanethiol) to produce a separated binary component SAM (Bumm et al., 1999a). It was initially demonstrated that these two alkanethiolates were similar enough in chain-length that they were miscible on a surface and are randomly mixed when codeposited from a 95% decanethiol/5% dodecanethiol solution (Figure 5). In this figure, the protruding features are attributed to dodecanethiolate molecules. It is important to note that the longer dodecanethiolate molecules are not adsorbing at domain boundaries or near substrate defects in the films, as was observed with the amide-containing alkanethiolate SAMs studies. In contrast, there appears to be true randomness in the monolayers as to where the dodecanethiolate molecules are adsorbing.
To ascertain the effect of annealing of the SAMs, Bumm et al. formed a single component SAM of dodecanethiolate on Au{111}. This was then annealed at 78 °C in ethanol for 1 h, which succeeded in partially desorbing the monolayer (Figure 6). Annealing also decreased the defect density and structural domain boundaries which are normally found in alkanethiolate SAMs. Thus, 'clean' SAMs of dodecanethiolate could be produced with this process.
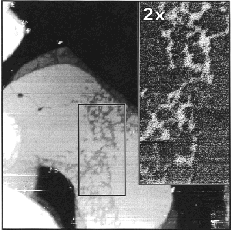
larger picture |
Figure 6. 5000 Å × 5000 Å STM image showing both reduced defect density and partial monolayer desorption after thermal annealing in neat ethanol. The inset is an apparent tunneling barrier height image (2x greater magnification) acquired over the area indicated by the rectangle. Imaging conditions: Vtip=+1 V, I=10 pA. |
It is known that alkanethiolate molecules undergo significant desorption at 60 °C(Finklea et al., 1993). Therefore, dry annealing processes that had been previously used to increase the domain size of alkanethiolate SAMs also introduced defects within the monolayer domains (Camillone et al., 1994). Since the annealing process is performed in solution, it is expected that as the SAM is heated, adsorbate molecules in the solution will undergo exchange processes more readily with the desorbing molecules due to the raised temperature.Also, increased temperatures will support lateral diffusion on the surface. Therefore, a situation that approaches equilibrium more closely than traditional SAMs formed at room temperature will occur.
After partial desorption of the initial single component monolayer, the substrate was immersed in a 1 mM ethanolic solution of decanethiol for 6 h at room temperature (Figure 7). This resulted in virtually defect-free islands of dodecanethiolate molecules surrounded by domains of decanethiolate molecules in which the defect density and structural domain boundaries were typical for that of SAMs adsorbed at room temperature.

larger picture |
Figure 7. A dodecanethiolate SAM was heated in a 1 mM decanethiol solution at 78 °C for 1 h, partially desorbing the SAM. Subsequent immersion in a 1 mM solution of decanethiol for 6 h at room temperature produced phase separated films. Typical STM image of a decanethiol/dodecanethiol SAM after post-adsorption processing. The arrows identify examples of three types of SAM boundaries: (a) Decanethiolate/dodecanethiolate laterally epitaxial boundaries and (b) Decanethiolate/dodecanethiolate boundaries associated with (c) Decanethiolate/decanethiolate structural domain boundaries. Imaging conditions: Vtip=+1 V, I=5 pA, 500 Å x 500 Å. |
These film preparations proved useful in quantifying the contributions of the molecules to electron transport as a function of chain length (Bumm et al., 1999b; Weiss et al., 1998; Arnold, 1997).
Along with the separation that is achieved using this annealing process, molecularly sharp domain boundaries are also apparent (Figure 8). The lattice structure between the regions of decanethiolate and dodecanethiolate is continuous; that is, there are no chain twisting or distinct physical defects at the domain boundaries. These boundaries are stable in air at room temperature. In contrast, domain boundaries between different regions of decanethiolate from the unprocessed SAM adsorption are distinct and indicate chain twist that is typical of domain boundaries in alkanethiolate SAMs (Figure 7, c). At the defect-free boundaries between decanethiolate and dodecanethiolate domains, the steps between the molecular terraces, the longer dodecanethiolate molecules are more accessible and thus can expose unique functionality on the film surface, possibly enabling selective deposition or reaction at such topologically one-dimensional sites.Therefore, another level of patterning can be realized in which the exposed longer-chain functional groups at the boundaries can be manipulated to undergo reactions at those specific sites.Post-adsorption processing (i.e., annealing) of SAMs proves useful as a way to control the type and density of defects in SAMs so that patterning following adsorption can be accurately controlled.

larger picture |
Figure 8. 250 Å x 250 Å STM image showing molecularly sharp boundaries at the edges of the areas covered by decanethiolate and those by dodecanethiolate. Note the lateral epitaxy and lack of physical defects at the domain edge. Imaging conditions: Vtip=+1 V, I=10 pA. |
Controlling Molecular Placement by Insertion
Whereas codeposition of a multi-component SAM has been demonstrated in controlling the formation of nanometer-scale domains, it is also possible to control the insertion of individual molecules into an alkanethiolate matrix. The insertion process that we have studied involves a family of molecules known as 'molecular wire' candidates. These molecules typically are linear conjugated oligomers with a phenylene ethynylene backbone. Measurements of the bulk conductivity of this family of molecules has been obtained (Zhou et al., 1997); however, for molecular devices it is necessary to know whether the conductivity is due to a large number of these molecules or if they conduct as single molecules.
In order to achieve a quantitative, non-ensemble measurement of the conductivity of these molecules, isolation of the molecular wires in a matrix of a less-conductive alkanethiolate SAM was performed (Bumm et al., 1996). Because of its lateral resolution, STM could then be used to probe the conductivity of individual matrix-isolated molecules. Bumm et al. showed that these types of measurements were possible using STM and the insertion process.
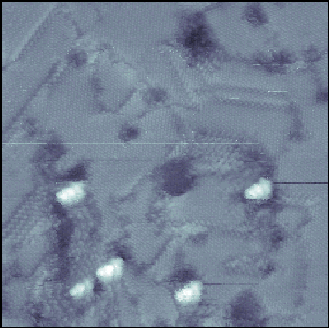
larger picture |
| Figure 9. 350 Å x 350 Å STM image of a molecular wire inserted into a dodecanethiolate SAM. The wires (topographically higher spots) insert at domain boundaries and substrate defects. Imaging conditions: Vtip= +1 V, I = 1 pA. |
Cygan et al. studied the insertion process of these molecules (Cygan et al., 1998). Two types of films were analyzed: (1) films codeposited from solutions of a thioacetate-protected molecular wire and dodecanethiol, and (2) films deposited for 18 h from solutions of dodecanethiol followed by immersion into a 0.3 mM solution of one of the molecular wires for 0.5 or 1 h. (Films formed from a 0.3 mM solution of one of the molecular wires were also analyzed, but did not demonstrate any significant order and were not crucial to this study.) The films were characterized by ellipsometry, contact angle measurements, and infrared spectroscopy. Additionally, the films were analyzed using STM to obtain images of the outcome of the insertion procedure.
When codeposited films of molecular wire candidates and alkanethiolates were prepared, they were found to be highly disordered compared to pure alkanethiolate films. Even at low concentrations of the molecular wire in solution (11%), no crystalline order was observed.It was concluded that the molecular wires disturb the ordering process during deposition.
However, the insertion process allows us to produce well-ordered SAMs before exposure to the molecular wires in solution. Prior to and immediately following insertion, the crystalline order of the dodecanethiolate matrix could easily be observed (Figure 9). Substrate defects and domain boundaries that are indicative of alkanethiolate SAMs also remain before and after insertion. After insertion, the films are interspersed with protrusions appearing ~4-6 Å higher than the film in STM images. These were attributed to the molecular wires, as they were not present in the films imaged immediately before insertion. The molecular wires insert at domain boundaries, substrate defects, and step edges. It is thus assumed that the molecular wire candidates are chemisorbing to the Au surface through a S-Au bond at sites that are particularly accessible to the molecules.
This same insertion strategy has been used to bind tethers designed and synthesized by Weck and Grubbs to serve as the anchor and initiation points for ring-opening metathesis polymerization (ROMP) (Weck et al., 1999; Weck et al., 1998). By controlling the initial SAM defect density, we controlled the number of inserted tethers and their separation. Polymerization from these norbornene-terminated phenylene ethynylene thiolates then resulted in individual, isolated polymer chains in the low SAM defect limit, or microns-wide polymer brushes where large defects had been opened to allow substantial insertion. Since ROMP yields a living polymer, the chain length could be controlled via monomer supply and polymerization time.
The insertion process is an effective way to measure single molecules. Further control over the placement of the molecules can be achieved by controlling the substrate defect density as in solution annealing (detailed in the above section), or by carefully selecting the matrix molecule to control the uniformity of the environment of the inserted molecules (Charles, 1999).
Conclusions
Organic monolayers can be patterned through phase separation due to terminal and internal functionality, post-adsorption processing leading to separation, and controlled placement of individual molecules through insertion. The key to each of these processes is controlling intermolecular interactions and defect type and density so as to control the film dynamics. Ongoing research in our group includes studying possible phase separation due to differing head groups and substantial differences in chain-lengths of the alkanethiolate molecules.
The methods detailed here have been applied to control the placement of molecules. However, for these to be useful, it will be necessary to dictate precisely where individual molecules are placed. By simultaneously controlling substrate, film, and subsequent reactivity, this may become possible.
Acknowledgements
We would like to acknowledge the contributions to this work of our highly productive and stimulating collaborations with Dave Allara, Bob Grubbs, Jim Hutchison, Jim Tour, and Marcus Weck. We thank the Army Research Office, Defense Advanced Research Projects Agency, National Science Foundation, and Office of Naval Research for support of this work.
References
Allara, D. L. (1995) Biosens. Bioelec., 10, 771-783.
Arnold, J. J. (1997) M.S. Thesis, Department of Chemistry, The Pennsylvania State University.
Bain, C. D.; Evall, J.; Whitesides, G. M. (1989) J. Am. Chem. Soc., 111, 7155-7164.
Bain, C. D.; Whitesides, G. M. (1988) J. Am. Chem. Soc., 110, 6560-6561.
Bain, C. D.; Whitesides, G. M. (1989a) J. Am. Chem. Soc., 111, 7164-7175.
Bain, C. D.; Whitesides, G. M. (1989b) Langmuir, 5, 1370-1378.
Becker, R. S.; Golovchenko, J. A.; Swartzentruber, B. S. (1987) Nature, 325, 419-421.
Binnig, G.; Quate, C. F. ; Gerber, C. (1986) Phys. Rev. Lett., 56, 930-933.
Binnig, G.; Rohrer, H.; Gerber, C.; Weibel, E. (1982) Appl. Phys. Lett., 40, 178-180.
Boulas, C.; Davidovits, J. V.; Rondelez, F.; Vuillaume, D. (1996) Phys. Rev. Lett., 76, 4797-4800.
Bumm, L. A.; Arnold, J. J.; Charles, L. F.; Dunbar, T. D.; Allara, D. L.; Weiss, P. S. (1999a) J. Am. Chem. Soc., 121, 8017-8021.
Bumm, L. A.; Arnold, J. J.; Dunbar, T. D.; Allara, D. L.; Weiss, P. S. (1999b) J. Phys. Chem. B, 103, 8122-8127.
Bumm, L. A.; Arnold, J. J.; Cygan, M. T.; Dunbar, T. D.; Burgin, T. P.; II, L. J.; Allara, D. L.; Tour, J. M.; Weiss, P. S. (1996) Science, 271, 1705-1707.
Camillone, N.; Eisenberger, P.; Leung, T. Y. B.; Schwartz, P.; Scoles, G.; Poirier, G. E.; Tarlov, M. J. (1994) J. Chem. Phys., 101, 11031-11036.
Charles, L. F. (1999) M.S. Thesis, Department of Chemistry, The Pennsylvania State University.
Chen, J.; Reed, M. A.; Rawlett, A. M.; Tour, J. M. (1999) Science, 286, 1550-1551.
Clegg, R. S.; Hutchison, J. E. (1996) Langmuir, 12, 5239-5243.
Clegg, R. S.; Hutchison, J. E. (1999) J. Am. Chem. Soc., 121, 5319-5327.
Clegg, R. S.; Reed, S. M.; Smith, R. K.; Barron, B. L.; Rear, J. A.; Hutchison, J. E. (1999) Langmuir, 15, 8876-8883.
Clegg, R. S.; Smith, R. K.; Hutchison, J. E. In preparation.
Collet, J.; Tharaud, O.; Chapoton, A.; Vuillaume, D. (2000) Appl. Phys. Lett., 76, 1941-1943.
Collet, J.; Vuillaume, D. (1998) Appl. Phys. Lett., 73, 2681-2683.
Cygan, M. T.; Dunbar, T. D.; Arnold, J. J.; Bumm, L. A.; Shedlock, N. F.; Burgin, T. P.; Jones, L.; Allara, D. L.; Tour, J. M. ; Weiss, P. S.(1998) J. Am. Chem. Soc., 120, 2721-2732.
Dubois, L. H.; Nuzzo, R. G. (1992) Ann. Rev. Phys. Chem., 43, 437-463.
Eigler, D. M.; Schweizer, E. K. (1990) Nature, 344, 524-526.
Finklea, H. O.; Ravenscroft, M. S.; Snider, D. A. (1993) Langmuir, 9, 223-227.
Folkers, J. P.; Laibinis, P. E.; Whitesides, G. M.; Deutch, J. (1994) J. Phys. Chem., 98, 563-571.
Gimzewski, J. K.; Joachim, C. (1999) Science, 283, 1683-1688.
Hla, S.-w.; Bartels, L.; Meyer, G.; Rieder, K.-H.(2000) Phys. Rev. Lett., 85, 2777-2780.
Hong, S.; Zhu, J.; Mirkin, C. A. (1999) Science, 286, 523-525.
Lewis, P.A.; Smith, R. K.; Kelly, K. F.; Bumm, L. A.; Reed, S. M.; Clegg, R. S.; Hutchison, J. E.; Weiss, P. S. In preparation.
Liu, G.-Y.; Xu, S.; Qian, Y. (2000) Acc. Chem. Res., 33, 457-466.
Poirier, G. E. (1997) Chem. Rev., 97, 1117-1127.
Rai-Choudhury, P. (1997) Handbook of Microlithography, Micromachining, and Microfabrication (London: SPIE Optical Engineering Press) 768.
Smith, R. K.; Reed, S. M.; Lewis, P. A.; Monnell, J. D.; Clegg, R. S.; Kelly, K. F.; Bumm, L. A.; Hutchison, J. E. ; Weiss, P. S. J. Phys. Chem. B, in press.
Stranick, S. J.; Atre, S. V.; Parikh, A. N.; Wood, M. C.; Allara, D. L.; Winograd, N. ; Weiss, P. S. (1996) Nanotechnology, 7, 438-442.
Stranick, S. J.; Parikh, A. N.; Tao, Y.-T.; Allara, D. L.; Weiss, P. S. (1994) J. Phys. Chem., 98, 7636-7646.
Ulman, A. (1991) An Introduction To Ultrathin Organic Films: From Langmuir-Blodgett To Self-Assembly (San Diego: Academic Press, Inc) 442.
Ulman, A. (1996) Chem. Rev., 96, 1533-1554.
Weck, M.; Jackiw, J. J.; Rossi, R. R.; Weiss, P. S.; Grubbs, R. H. (1999) J. Am. Chem. Soc., 121, 4088-4089.
Weck, M.; Jackiw, J. J.; Weiss, P. S.; Grubbs, R. H. (1998) Proc. Poly. Mat., Sci., Eng., 79, 72-73.
Weiss, P.S.; Bumm, L. A.; Dunbar, T. D.; Burgin, T. P.; Tour, J. M.; Allara, D. L. (1998) Ann. NY Acad. Sci. 852, 145-168.
Weiss, P. S.; Eigler, D. M. (1993) NATO ASI Series E: Applied Sciences 235, 213-217.
Xia, Y.; Whitesides, G. M.(1998) Angew. Chem., Intl. Ed. Eng., 37, 550-575.
Zhao, X. M.; Xia, Y.; Whitesides, G. M. (1997) J. Mater. Chem., 7, 1069-1074.
Zhou, C.; Deshpande, M. R.; Reed, M. A.; Jones, L.; Tour, J. M.(1997) Appl. Phys. Lett., 71, 611-613.
|