Presenters

Brian Kennedy, Buck Institute
Dr. Kennedy earned his PhD from the Massachusetts Institute of Technology and is well known for his work during his graduate studies with Leonard Guarente, PhD, which led to the discovery that sirtuins (SIR2) modulate aging. He performed postdoctoral studies at the MGH Cancer Center associated with Harvard Medical…

Joris Deelen, Max Planck Institute for Ageing
Joris Deelen is leading the research group on Genetics and Biomarkers of Human Aeging at the Max Planck Institute for Aeging Research. The group studies the genetic mechanisms underlying healthy ageing in humans by investigating the effect of genetic variants that are unique to long-lived families on the…

Lynne Cox, Oxford University
Lynne Cox is the George Moody Fellow and Tutor in Biochemistry at Oriel at the University of Oxford. Her lab studies the molecular basis of human ageing, with the aim of reducing the morbidity and frailty associated with old age. Her lab studies progeroid Werner’s syndrome (WS), where cell senescence and early onset of many…
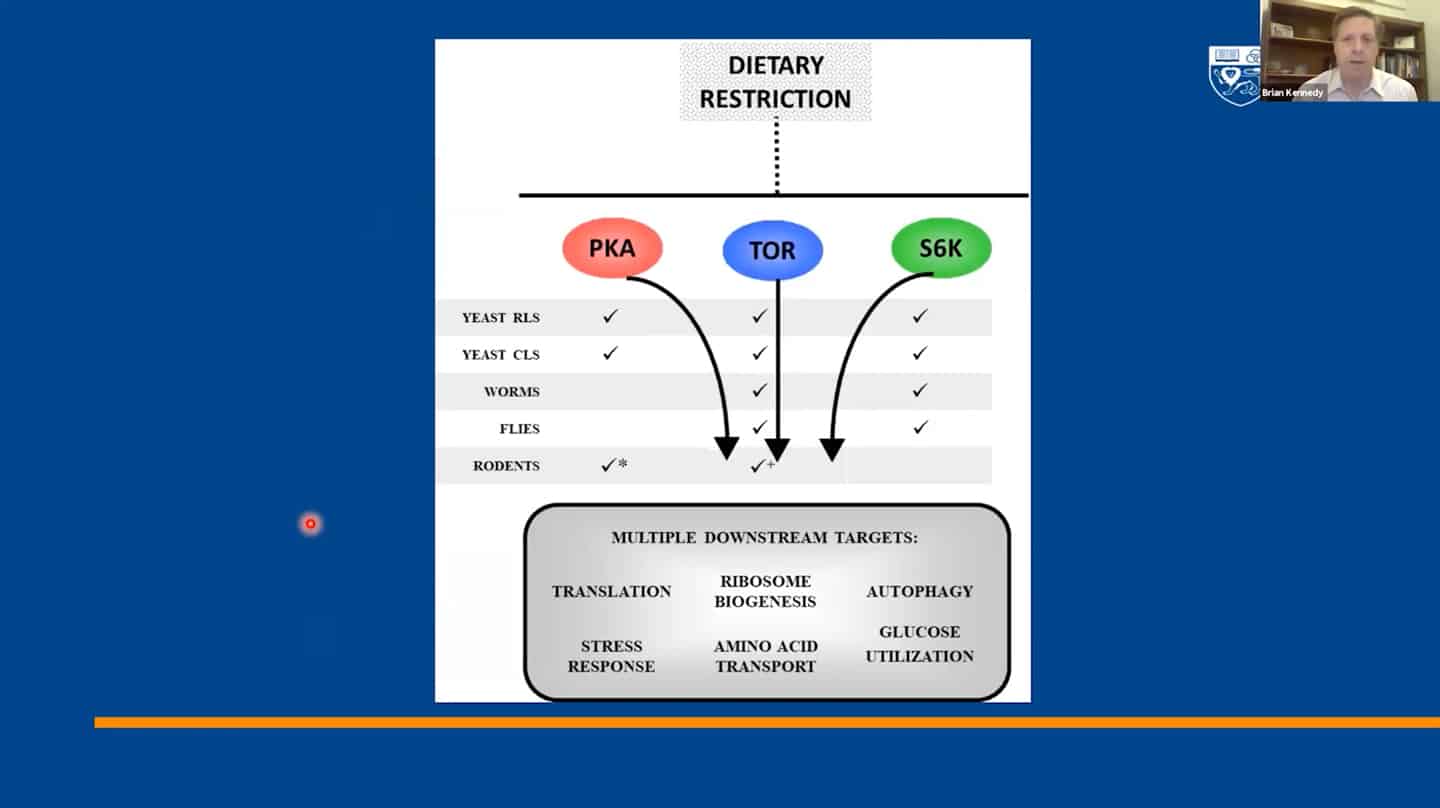
- We identified 300+ genes via basic research on invertebrates which served as a basis for most of the current research, but now basic research and model organisms are kind of overlooked in terms of funding and focus. It’s great that private money is entering the longevity space, but that seems to generate this unfortunate idea that basic research is something that’s no longer needed and not a priority. And yet we don’t even know how the 300+ genes regulating aging in yeast interact with each other, which ones regulate which pathways, how they coordinate aging, etc. Single-cell organisms like yeast may be the only way to answer this question.
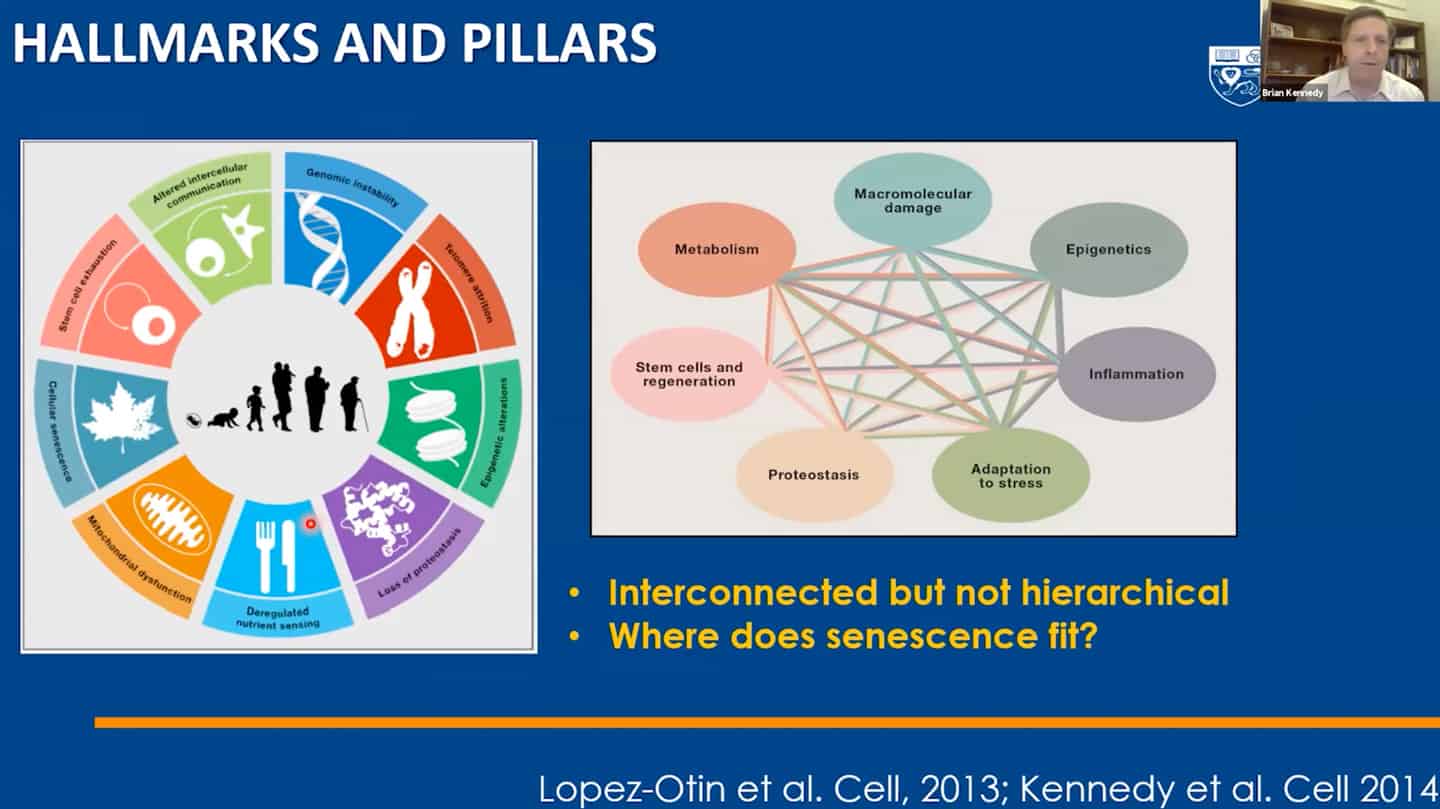
- We managed to categorize aging in pillars of aging / hallmarks of aging, but we still don’t understand the interaction between hallmarks of aging – that’s another example of open questions that are not being addressed.

- A lot of small molecules are now funded by companies, but a lot of them are still not and we are continuously discovering more.

- Roadmap to get to humans.

- Three different strategies for companies on how to get to humans.
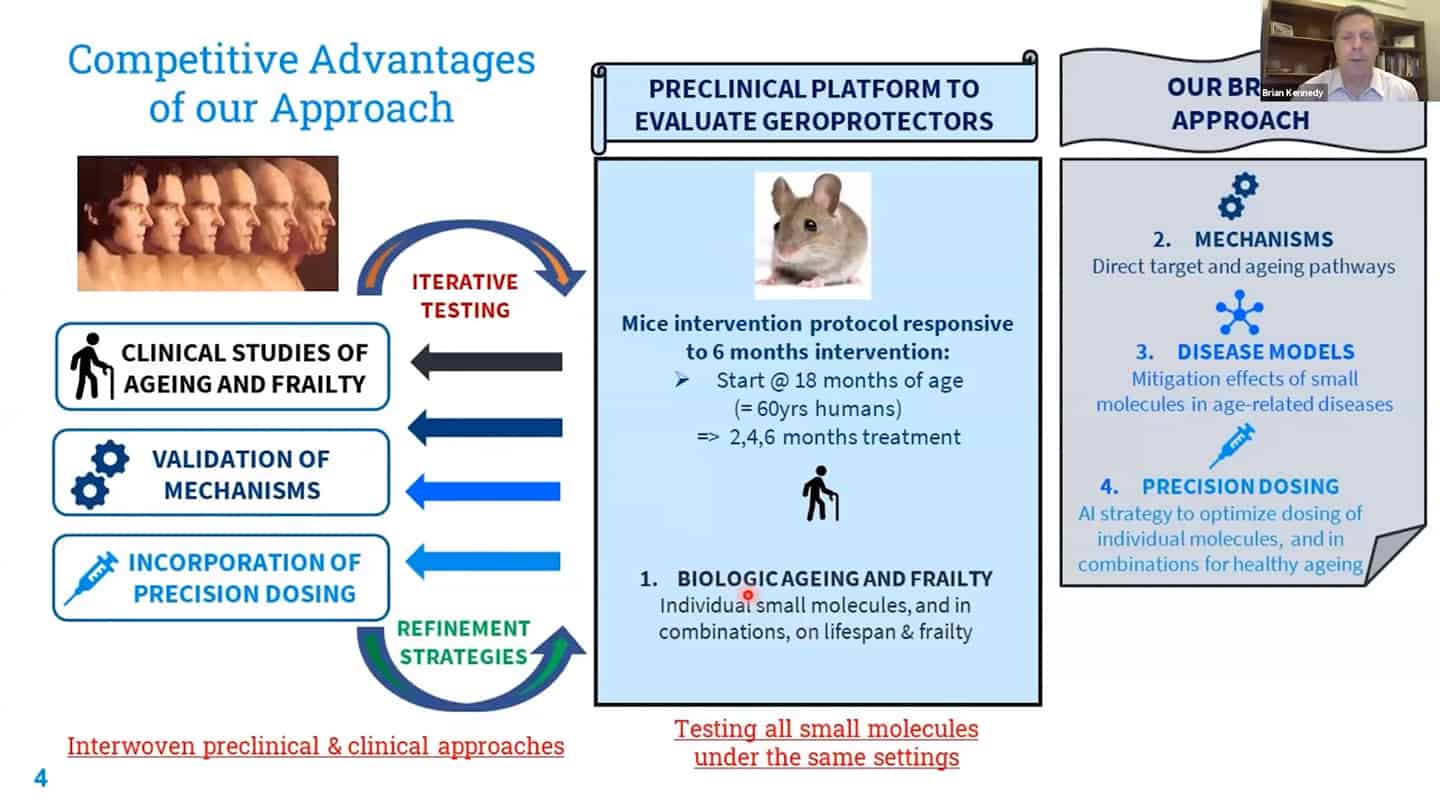
- We are working on recalibration of mouse studies, so that they are aligned with these shorter-time biomarker studies in humans, so we can work back and forth between humans and mice using validated biomarkers.

- Example of a study we’re running. Trying to test up to 10 of these molecules so we can contrast and compare how different interventions affect aging.

- We are using a number of different biomarkers for these studies, but there is the problem that we don’t know how aging biomarkers interact with each other.
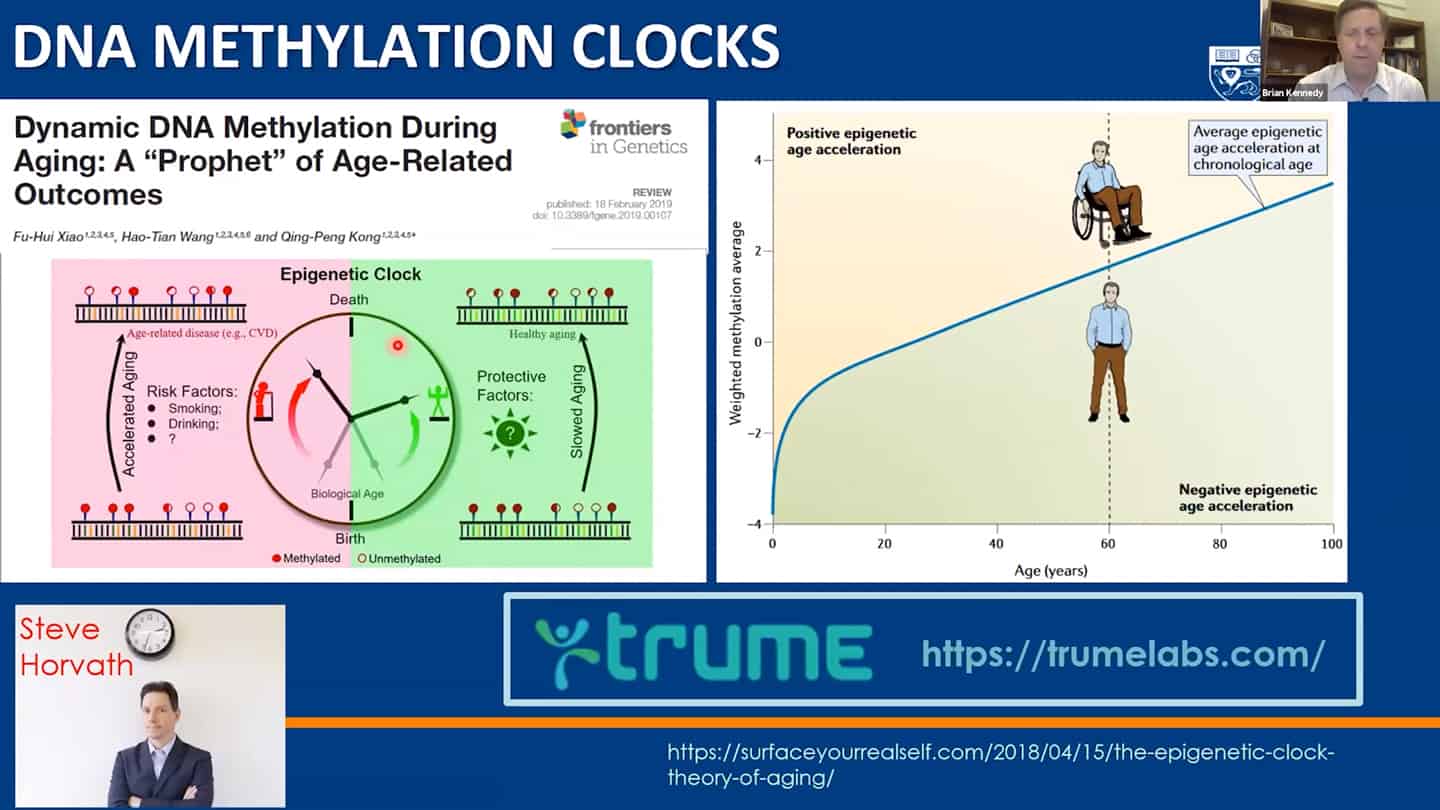
- Methylation clocks are commonly used, but even there are different methylation clocks, with different parameters, etc.
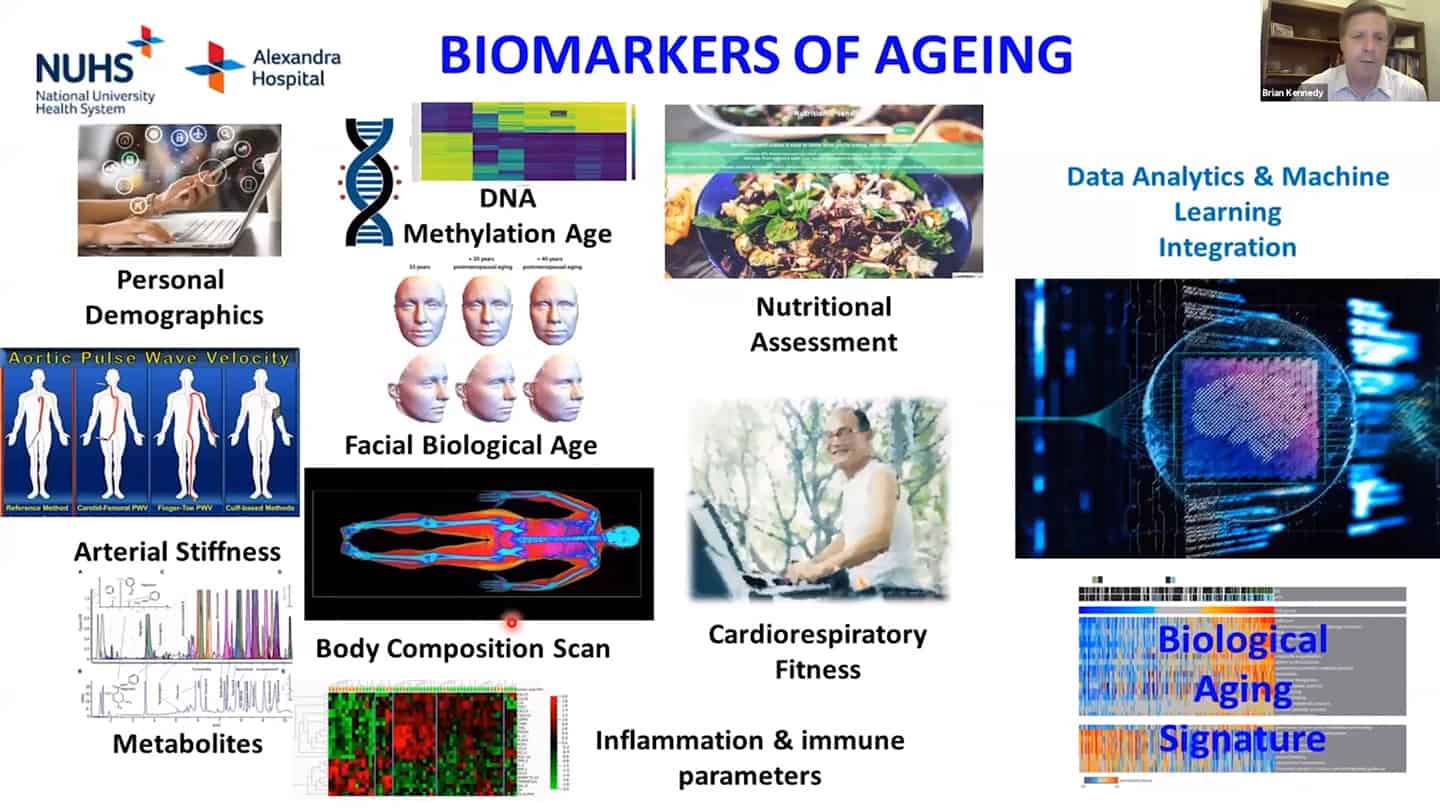
- We need to combine these interventions together, as well as measurements of the hallmarks together and validate what works in humans.

- Agree with Brian that we need to tie things together in the hallmarks of aging space, we think we know quite a bit about it and we need more, but there are some other things that we simply don’t know enough at all.
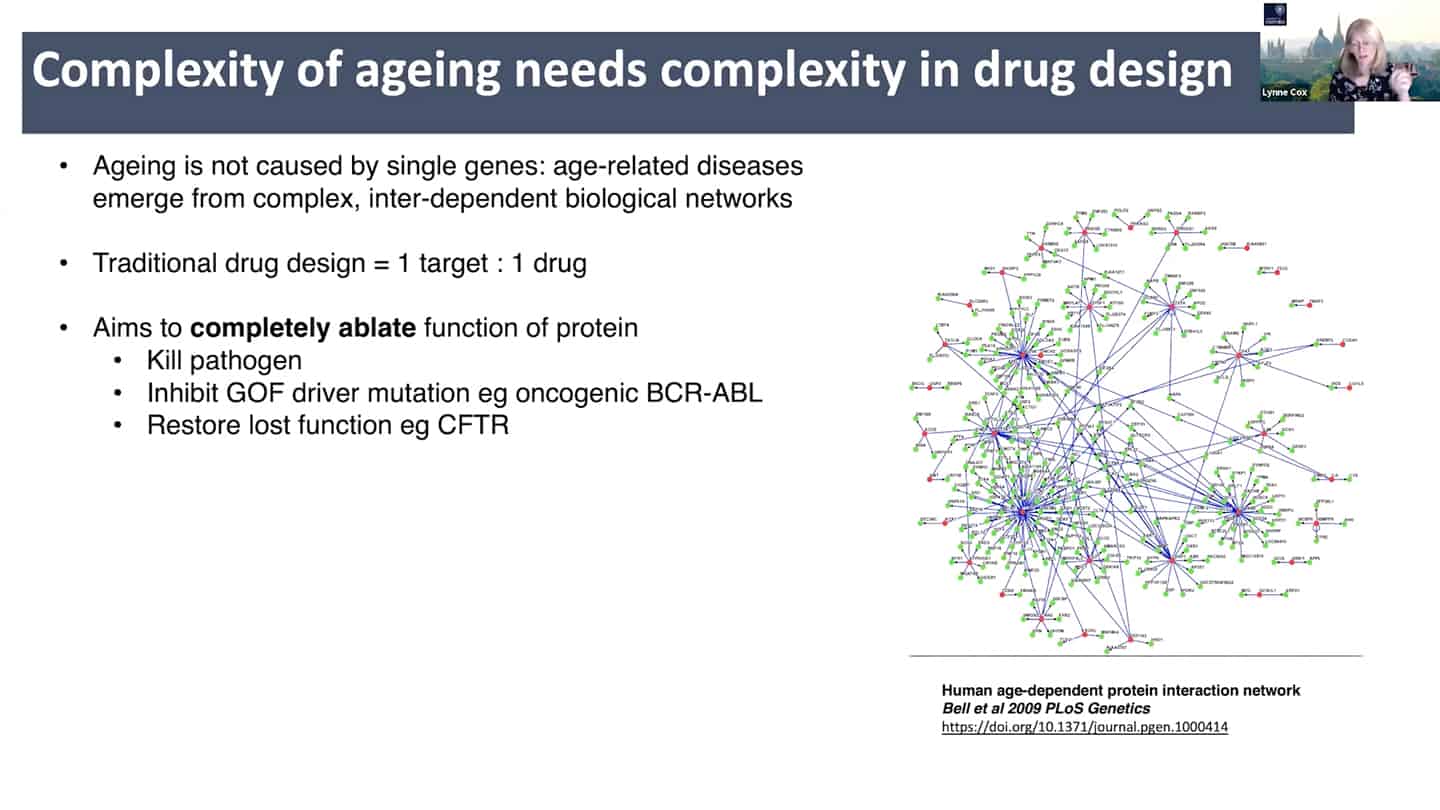
- New drug discovery approaches are needed. Aging is a complex inter-dependent and emergent network. Traditional approach is 1 target = 1 drug, where you try to annihilate the target completely to totally ablate it’s function. This does not work well with aging because of the interconnectedness and redundancy in aging pathways. There is probably broad agreement on this, but it is insufficiently stressed, perhaps because ultimately getting different (perhaps multi-target) interventions past FDA/EMA has historically been unusual outside of oncology and diseases like tuberculosis.
- See papers: Limits of the classic 1 knockout at a time methodology, Genetics of extreme human longevity to guide drug discovery for healthy ageing

- Current pharma philosophy of inhibiting target completely is like a hammer to fix a clog on a wristwatch, we need to find better solutions like modulating and damping down multiple nodes, rather than killing them completely, akin to fine tuning and tweaking wristwatch with tweezers instead of a hammer.

- This requires systems biology and drug synergy approaches – polypharmacology. Instead of 1 target = 1 drug, develop X targets = 1 drug approach, where one drug hits multiple targets and is target agnostic. And that approach requires function first phenotypic drug screening.
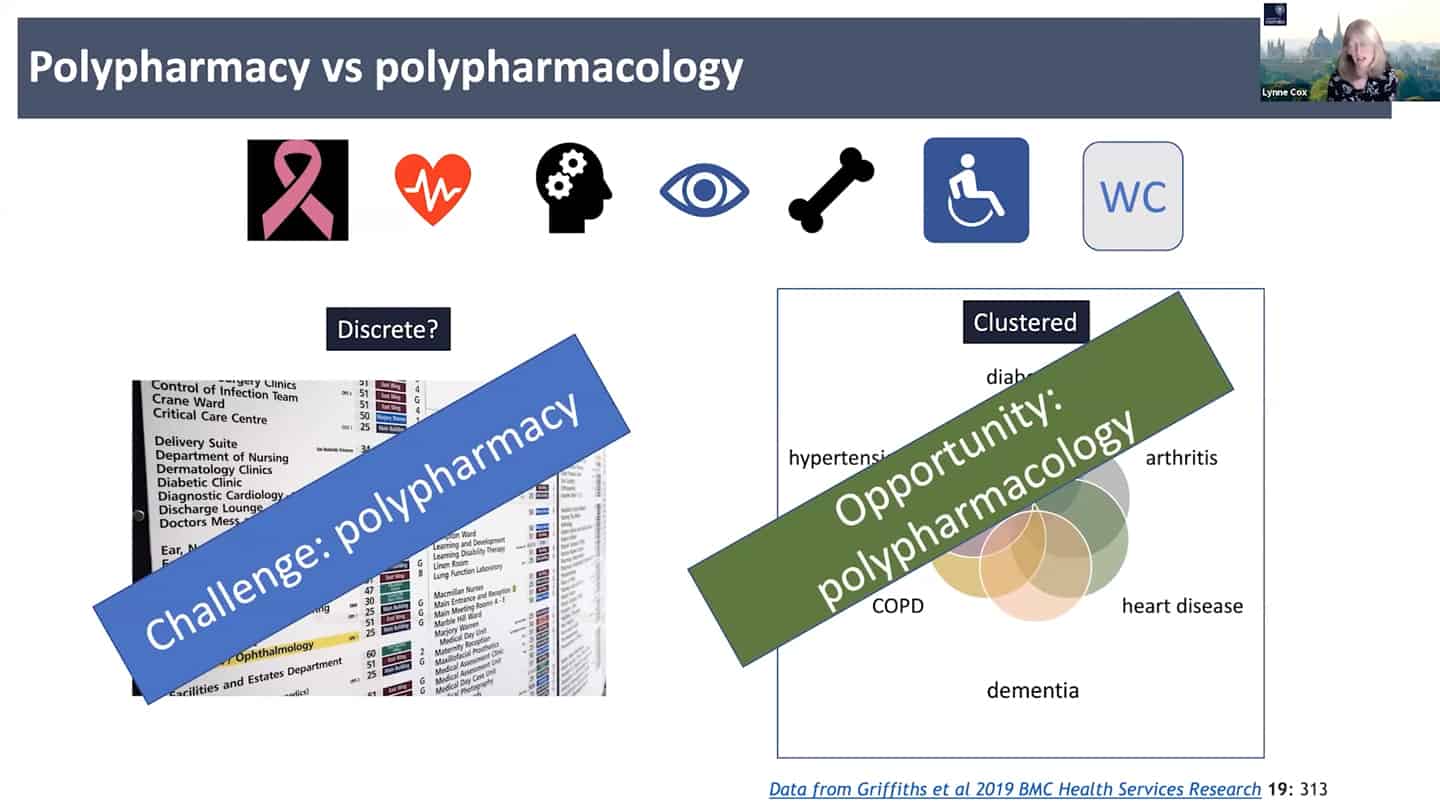
- Right now the standard approach is polypharmacy, for every problem a different expertise and clinic, instead of looking at the common denominators. We need to look at the shared clustered reasons, like aging, where we can hit the core of it all.
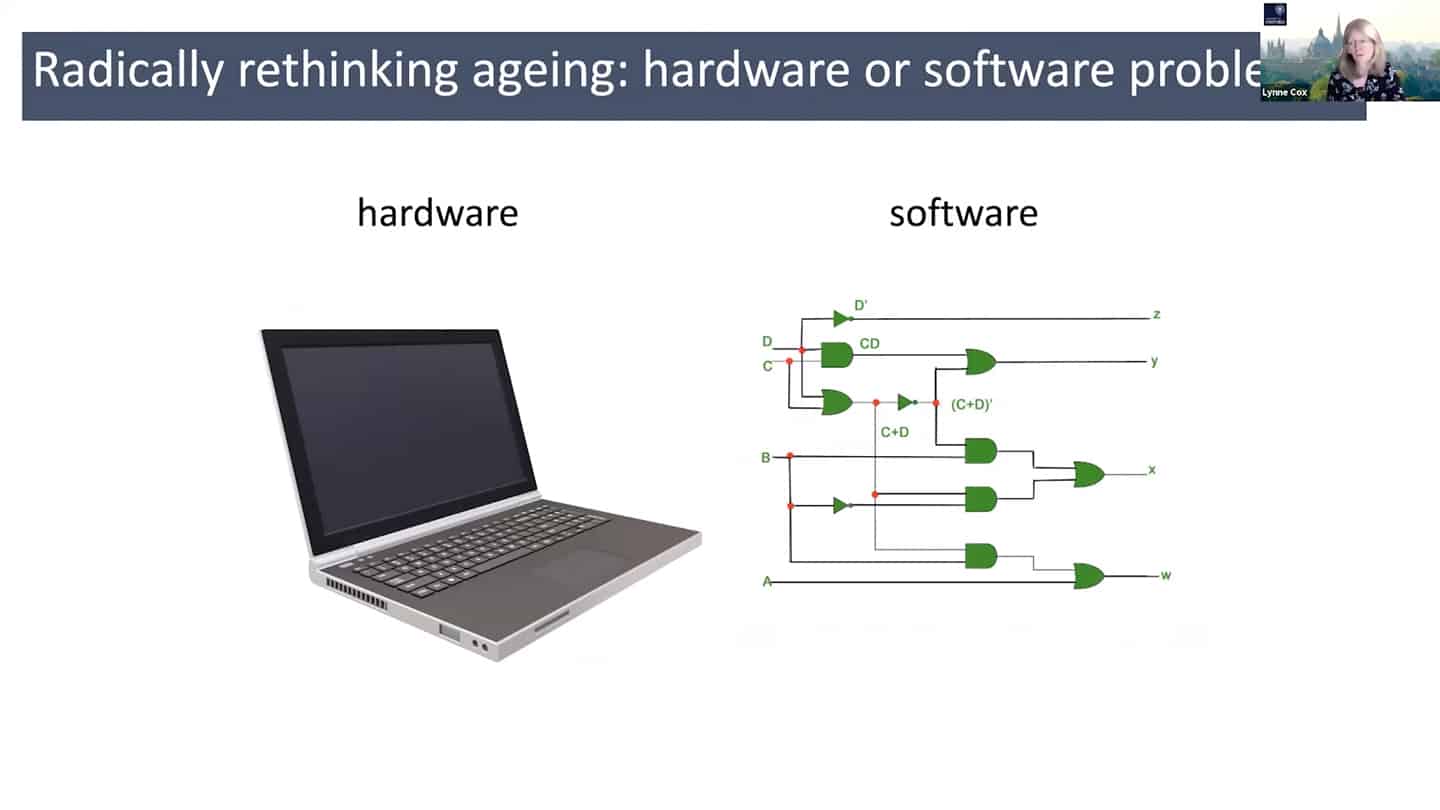
- In order to do all that, we need a different understanding of aging – maybe radically rethinking aging as a hardware and software problem could be an useful framework.
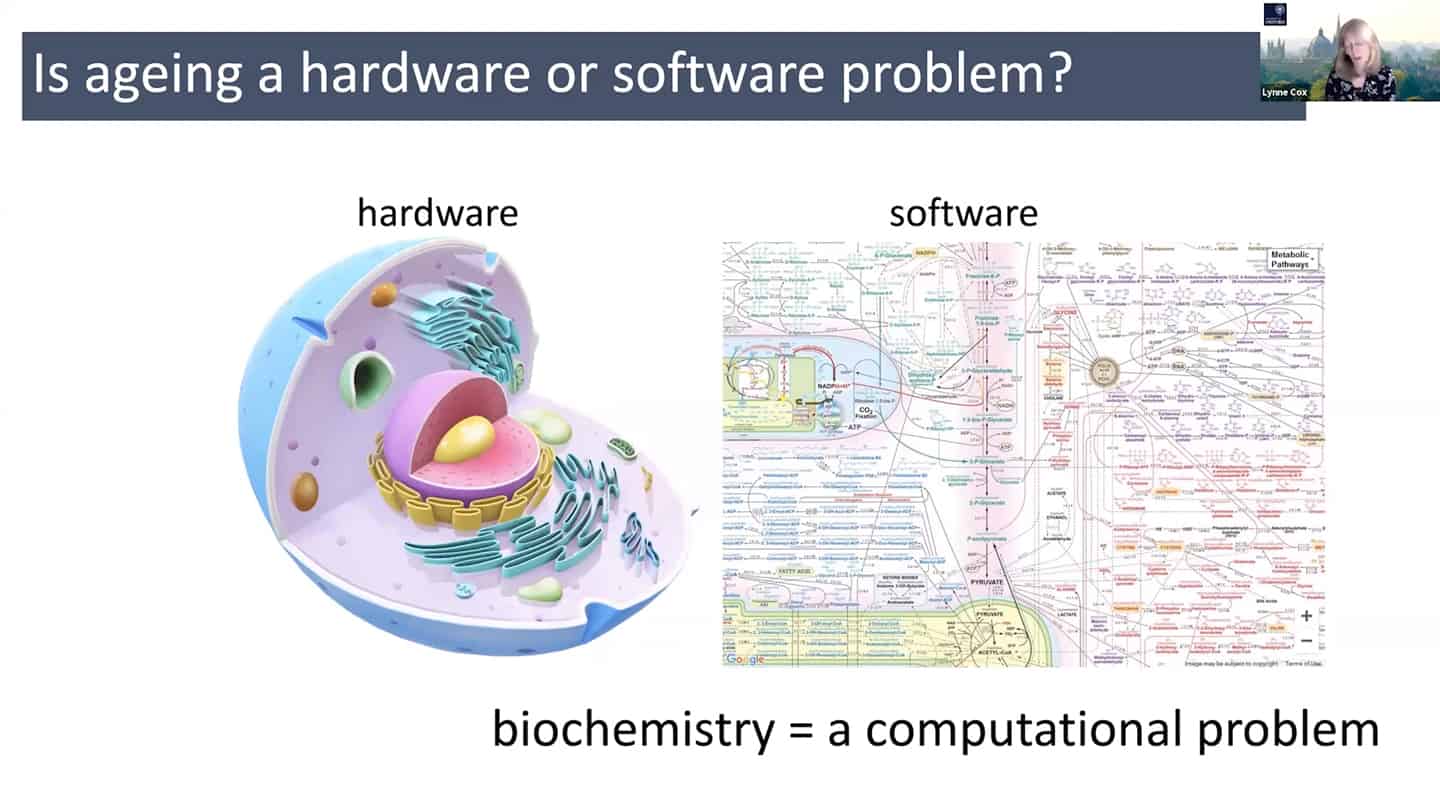
- In quite a reductionist sense, hardware is essentially a cell, organelles or macromolecules, while the software is the way that those things talk to each other, how the information flows through the system through biochemical pathways. And so we can think about biochemistry as a computational problem, and maybe as we accumulate enough data, we could plug it into a model and start to mimic aging in silico.

- mTOR is a good example – we know that young cells have correct mTOR programming that switches on and off, but in old senescent cells it is switched on all the time. So we can start to think about it in a computational way as a logic gate. In the young cells, it is a correctly working AND gate, and in the old cells, it is incorrectly working OR gate. So all we have to do is to go there and debug the software.
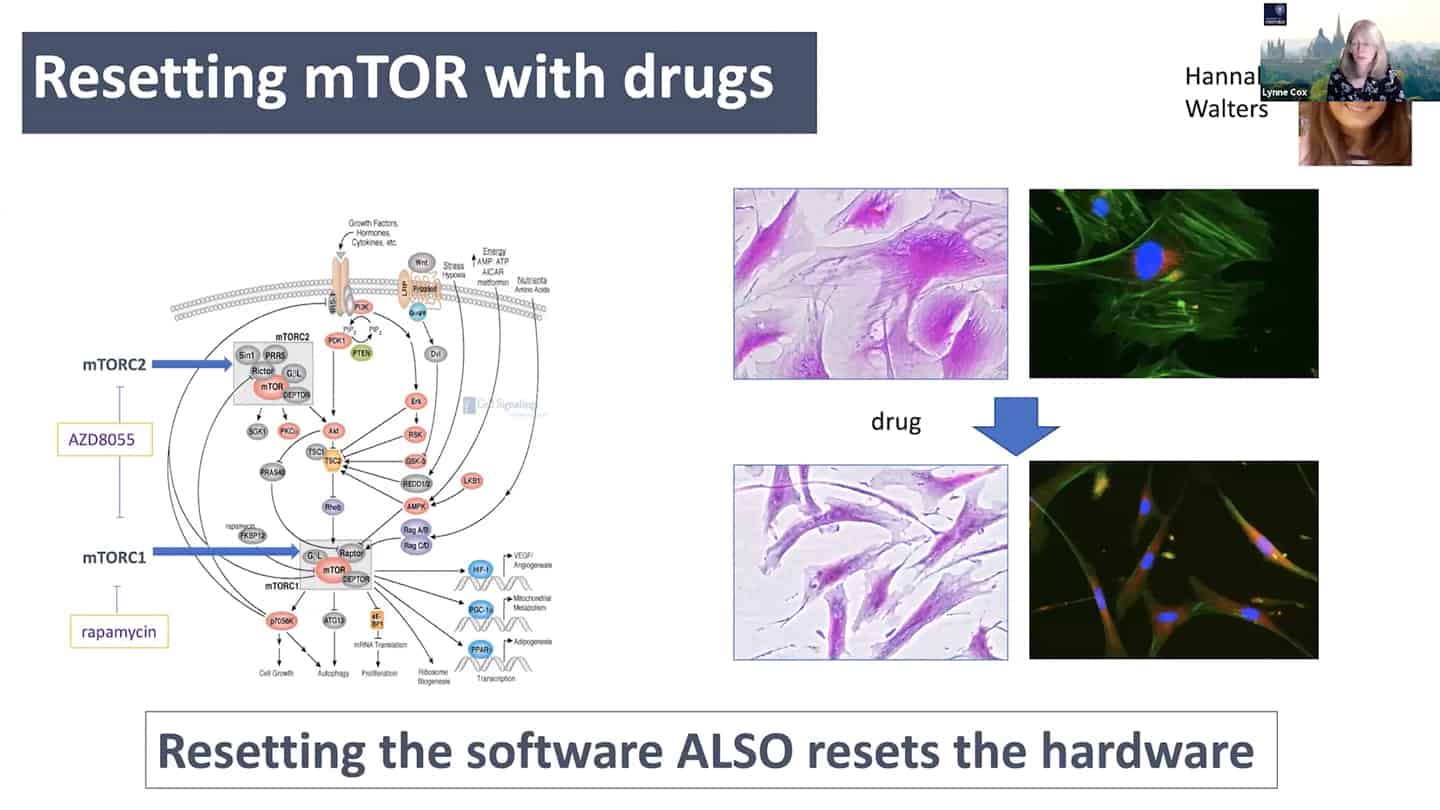
- As a proof of concept, we tried to reset the mTOR with a pan-mTOR inhibitor to see whether we can reset the software, and interestingly resetting the software did reset the hardware as well.

- We know that we don’t know how these genes interact with each other, how the cell components in these systems interact with tissues, organs, and systems, with microbiome, and even more how environmental factors affect all of that, both generationally and intergenerationally. And so what we need are biomarkers, we need to know what’s going on, we need full biochemical workup, systems modelling, and we also need big data. Not just biology -omics (geromics for gerontology), but also other factors like socioeconomic data (e.g. Open Life Data Framework).