Presenters

Christopher Leptak, CDER Biomarker Qualification Program, FDA
Christopher Leptak, Director of the CDER Biomarker Qualification Program at the FDA, will join us for a discussion on how to develop biomarkers for FDA-recognized diseases. While aging is not an FDA-recognized disease, and thus falls outside of the scope of this discussion, Christopher may help us shed light on the…


- With all the digital biomarkers popping up, the definition is becoming a little bit fuzzy in the public discourse, but the FDA has clear criteria for biomarkers.

- Important resource called BEST – long-term effort between NIH and FDA to collect a glossary and codify terminology within the biomarker space. It’s a living document that’s updated periodically trying to ensure that everybody can be on the same page.

- We try to define biomarker classes around their use as a way to differentiate them between each other – the 7 classes are listed above.

- But it’s probably easier to think about different types of biomarkers through this flow diagram.
- “Normal” physiology – For a given individual, a normal doesn’t mean unchanged, it means within defined parameters. For a given person, throughout the course of a day, or a period of time, depending on the characteristic you are looking at, it might change. And that normal variability is important, because if you wanna say that change is important and wanna make a regulatory action on it, you need to know what the level of change is with respect to normal variability. So within the normal states some of the biomarkers we are looking at are susceptibility or risk where you try to predict a likelihood of disease in the future even though at present you might not have clinically manifested signs or symptoms.
- Pathologic changes – The changes from normal variability can over time result in pathologic changes.
- Altered physiology – Those pathologic changes can lead to altered physiology.
- Clinical disease – And the altered physiology is leading to clinical disease. The FDA approves drugs based on a disease, and that group of diseases also changes over time. Within a clinical disease, once you have that definition in play, there are few classes of biomarkers, namely diagnostic, monitoring and prognostic.
- Diagnostic can fall into two camps – 1) the biomarker itself can be necessary and sufficient for the diagnosis of a disease where you don’t need any other information (we call that the capital D), 2) but the majority of biomarkers are (lowercase d) that along with other information and biomarkers and clinical signs and symptoms as a “gestalt” define a clinical disease, or even a disease by exclusion. That’s certainly a relevant class of biomarkers to help your patient population for a clinical trial.
- Monitoring is particularly helpful for a condition that has some time element to it (chronic or not just acute but somehow active in a longer time span). In such cases you might want to monitor and look for the disease activity, since some diseases are going to wax and wane in nature, and you want to enroll populations that have active disease in the clinical trial so you see whether the therapy is going to mitigate that or not.
- Prognostic class is important because in a clinical trial we ultimately want to look for a clinical benefit. And so you want to go into your trial enrollment and enrich for patients that are likely to have that clinical event of interest within the time of the clinical trial. If the clinical event of interest might take 10 years, then the biomarkers are going to be helpful to enrich the patient population and maybe even look for an earlier event than that 10 year outcome.
- Change of physiology – When a therapy is in place we look at the remaining classes of biomarkers, ultimately here we want to show some evidence of engagement. Pharmacodynamic basically says that the therapy has some biological effect. But it doesn’t say that the effect is either beneficial or detrimental. So that can be proof of concept or looking at the mechanism of action of your product.
Predictive help to take a patient population and subcategorize them to say “this group with this particular feature may be more likely to have a positive effect or more likely to have a side effect and be a safety concern”.
Safety is another way we monitor the effect of a therapy on a patient, because we know that every drug, even the OTC drugs have some safety concerns.
- Non-progression or reversal – Ultimately what we’re trying to do is to change the physiology and the clinical course of disease so we no longer have progression or perhaps even have reversal. This is where we get into the response part of the spectrum, which themselves can become the endpoints, and which are measuring non-progression or reversal of the disease.
- Improved clinical benefit – And a subset of those that are predictive of clinical benefit are surrogates. Surrogates can become the basis for approval be it accelerated or traditional.

- At the bottom is an example of one of the qualifications of COU a few years ago.
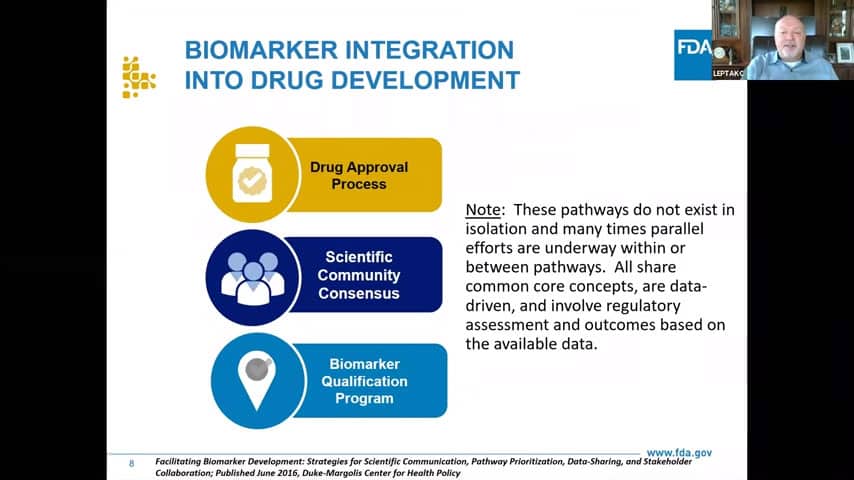
- Biomarkers come into the agency from many different places:
- Individual companies are bringing them under IND within the drug approval process, from where historically most of the biomarkers came and remains to be the biggest area.
- Then there is what we call the scientific community consensus – all the publications and scientific journals, professional societies and their position statements. This category is a great place for hypothesis testing, from where people can bring these hypotheses and bring them into either a drug approval process or a biomarker qualification program with all the evidence.
- Biomarker qualification program, which has been around now for over 15 years already, and has been formalised as a part of the 21st Century Cures Act in 2016.
- Do know that biomarkers are used and information lookup can come from each of these, we don’t have a preference. They are all data driven, on our side of the fence we have multidisciplinary teams that look at the information and many times biomarkers are developed in multiple categories simultaneously, which is helpful for us because we can find information from multiple sources.
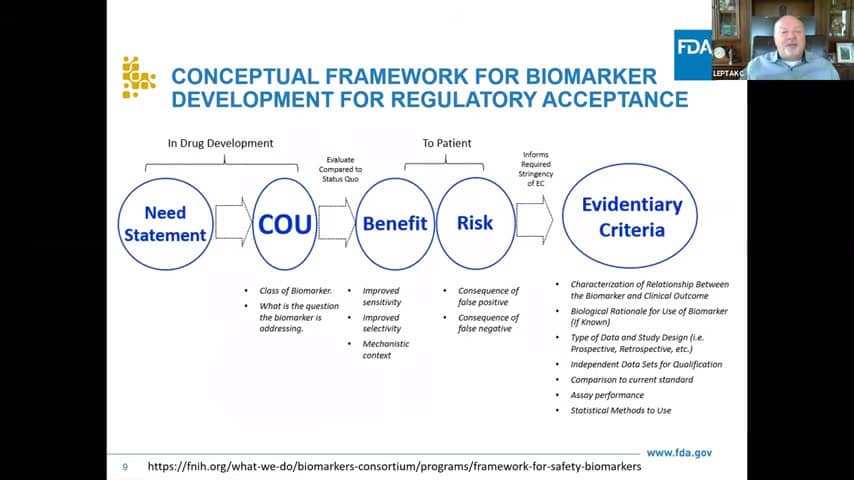
- Here are the elements of successful biomarker development for regulatory acceptance, taken from a paper that Allison mentioned, which we released so we can have better conversations about these elements to get you to your goal. We started with a need statement which for us is stated as drug development, which is sometimes confusing for folks trying to engage with us who are not trying to develop a drug themselves. Maybe they want to improve clinical care, which is a worthy goal, but not for our group, perhaps more for the devices group. So based on the Context of Use which we covered, we try to figure out what evidence we need to see based on COU and need, and we’re looking at benefits and risks for the patients, to help define the needed evidence. The evidence is going to include information around the analytics and the biomarker measurement, statistical analysis plan, in essence a fair bit of information that help support that biomarkers’ role.
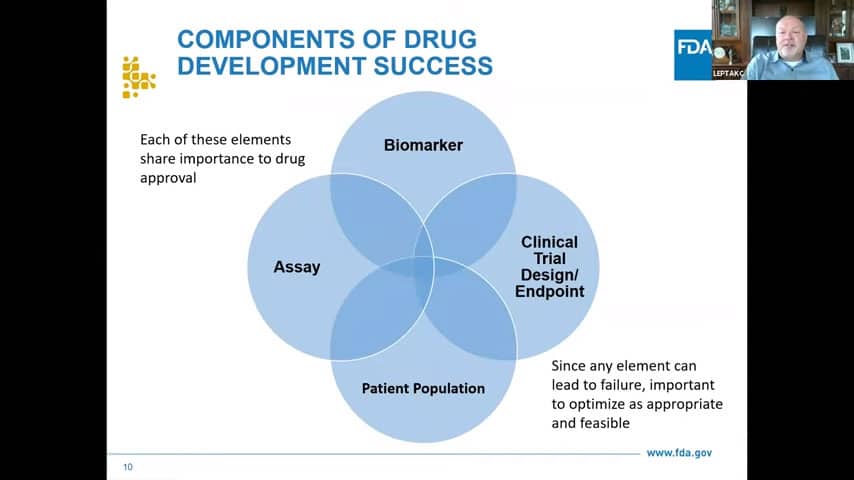
- Simplified, but many times where we actually get success is when all those elements overlap. So when you’re starting a new process, we encourage defining what your assumptions are in the first place and actually clearly writing them down. Because if you get a negative result, unless you really do your due diligence up front, you don’t know what really caused the negative result. Clearly defining your assumptions will help you interpret your results better, so at the end you know what exactly might have gone wrong.

- Analytics and measurement, we define them broadly into two separate buckets – analytical and clinical validation. Analytical validation establishes that you can measure something, but doesn’t mean that it means anything. Clinical validation means that the measurement has biological clinical meaningfulness. That’s what we’re looking for in clinical data to help support whatever it’s COU might be.

- 21th Century Cures put us in a proactive role and gave us a much more collaborative process with a better structure, targets and timelines. This gives you a way to build a plan that you can then execute – Letter of Intent, Qualification Plan, and Full Qualification Package.

- Transparent decision making letters, shared learning culture. If you are developing an imaging biomarker, you can check a lot of information from other biomarkers and discussions around them, because you can see the past decisions. Over time we see a great improvement in the quality of submissions because of this even with people that we didn’t engage with before that much.

- You do not submit primary data for LOI, this is relatively short 10-20 pages tops, very focused and summary level information.
- Qualification is the bottleneck usually, it’s where we do a deep dive in science provided. Here’s where you’re saying what you see in science today, what’s the goal, what are the steps to get to the goal and how I intend to address the gaps. And if we agree on that, you can execute the plan knowing that if the data is supportive, we will be happy and won’t ask for more things down the road.
- Full qualification plan is where you submit all the primary data, analysis and interpretation and we do our analysis and see if we come to the same conclusion or not.

- Internally we have a three tiered review: First tier is within the program, we look at whether it is administratively complete, if you’re telling a clear story. What we find most of the people do is that they try to put too much information. We want to have a targeted conversation about a specific idea, so you need to be able to tell a clear story that is focused on a specific thing. Once you have a good story to tell we take our subject matter experts, look at it in earnest, put together recommendations and at last take it to a committee and the discipline chiefs.

- People are always interested in surrogates – there is a table of surrogate endpoints that is updated every 6 months.

- Finally if you are a partner or have an active IND with us, there are these new type C meetings for surrogate endpoints so that is something you can explore.