
Advances last year in the bottom-up design and fabrication of increasingly complex atomically precise nanostrutures were so rapid we were not able to cover as many as we wanted. Before we dive into this year’s advances, we are catching up on some of the important advances we missed last year. Perhaps the most active area of molecular engineering research last year was the de novo protein design area, originally proposed by Foresight co-founder K. Eric Drexler as “a path to the fabrication of devices to complex atomic specifications”. Our most recent post in this area cites five earlier posts last year about protein design.
Continuing our coverage of important advances in protein design, the two co-winners of the 2004 Feynman Prize, Theory category (for developing the Rosetta software suite for biomolecular modeling and design) both reported important protein design advances in adjacent papers in Science last May. A perspective commentary “Inspired by nature” in the same issue by Ravit Netzer and Sarel J. Fleishman of the Weizmann Institute of Science points out that the great success over the past decade of de novo designing proteins that folded exactly as designed and were very stable has not produced all of the “important structural features seen in protein interfaces and enzyme active sites”. They note that computer algorithms like Rosetta used to design proteins optimize stability. “By contrast, evolution selects proteins for their ability to perform a vital molecular function, often at the expense of stability.” They discuss the complementary approaches to this issue taken by David Baker and his collaborators (today’s post) and by Brian Kuhlman and his collaborators (tomorrow’s post).
Writing in Geekwire, Alan Boyle reports “Scientists add twists to protein designs“:
Biochemists from the University of Washington have engineered complex protein molecules with additional chemical bonds that make it possible to mix and match them like the base pairs of DNA.
The designer proteins, described today in a paper published by the journal Science [abstract, full text PDF courtesy of Baker Lab], could open the way for a kind of synthetic coding system modeled after the groundbreaking double-helix DNA code system discovered by James Watson and Francis Crick back in 1953.
“Think of it this way: The principle of heredity is Watson-Crick base pairing between the two complementary strands of DNA. We invent in the paper an analogous pairing arrangement for proteins,” David Baker, director of the UW’s Institute for Protein Design, told GeekWire in an email.
Protein molecules can be folded into a wide variety of shapes, which help determine how they function in cells. A software platform called Rosetta was invented at the UW more than a decade ago to analyze protein-folding patterns. Rosetta, along with a more recently developed program called HBNet, played a key role in designing new breeds of protein molecules that include additional hydrogen bonds. …
As explained on the Baker Lab web site “De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity“:
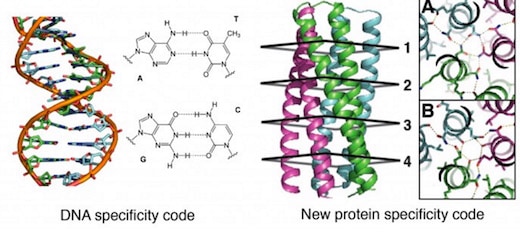
General design principles for protein interaction specificity are challenging to extract. In DNA, specificity arises from a limited set of hydrogen-bonding interactions in the core of the double helix to design and build a wide range of shapes. In proteins, specificity arises largely from buried hydrophobic packing complemented by irregular peripheral polar interactions. Protein-based materials have the potential for even greater geometric and chemical diversity, including additional functionality. Here we describe a general approach for designing a wide range of protein oligomers that have interaction specificity determined by modular arrays of extensive hydrogen bond networks. We use the approach to design dimers, trimers, and tetramers consisting of two concentric rings of helices, including previously not seen triangular, square, and supercoiled topologies. X-ray crystallography confirms that the structures overall, and the hydrogen-bond networks in particular, are nearly identical to the design models, and the networks confer interaction specificity in vivo. The ability to design extensive hydrogen-bond networks with atomic accuracy enables the programming of protein interaction specificity for a broad range of synthetic biology applications; more generally, our results demonstrate that, even with the tremendous diversity observed in nature, there are fundamentally new modes of interaction to be discovered in proteins.
Because hydrogen bond networks play much more complex and subtle roles in protein structures than they do in the simple molecular recognition networks that make DNA nanotechnology possible, Boyken et al. begin by developing a general computational method, HBNet, to rapidly list all side-chain hydrogen-bond networks possible with a given protein backbone structure. There are a large number of possibilities because there are more polar amino acids than there are DNA bases, and each amino acid side chain can adopt multiple conformations (rotameric states) depending on the protein backbone. HBNet computes hydrogen-bonding and steric repulsion interactions between all conformations of all pairs of polar side chains. It then identifies networks of three or more residues connected by hydrogen bonds with little steric repulsion. Networks are rejected if they contain buried polar groups that do not make hydrogen bonds.
To take advantage of networks of repeating structures to build scaffolds, they turned to coiled coils, specifically oligomeric structures with two concentric rings of helices. Systematic variation of helix parameters was used to generate a wide range of backbone structures. HBNet then searched tens of thousands of backbones to identify the small fraction that can support networks that can span the intermolecular interface.
A total of 114 dimeric, trimeric, and tetrameric designs spanning a broad range of superhelical parameters and hydrogen-bond networks were selected for experimental characterization. Genes encoding these protein structures were synthesized and expressed in bacteria. 101 of these expressed soluble proteins, which were purified for further characterization. 66 of these had the design oligomerization state, with tetramers having the lowest success rate (3 of 13 soluble designs assembled properly).
Proteins with two-ring designs were compared with corresponding structures having only the inner ring of helices. X-ray crystallography was used to identify those proteins with structures nearly identical to the design goals. In the most successful designs, nearly all buried polar groups made hydrogen bonds.
To test the role of the hydrogen-bond networks in conferring specificity for assembly into oligomers, control designs were made using the same backbones, but without HBNet. These control designs yielded only hydrophobic interfaces. These control designs were calculated to be energetically more favorable, but when expressed, the proteins were less soluble than their hydrogen-bond network counterparts, and those that were soluble enough to purify yielded multiple higher-molecular-weight aggregates instead of the design oligomers. The authors conclude from this and other experiments that designs in which hydrogen bond networks partition hydrophobic interface area into relatively small areas are more specific than designs with large contiguous hydrophobic patches. The best designs had hydrogen bond networksspanning the entire oligomeric interface, with each helix contributing at lest one side chain.
An additional set of trimers was designed with identical backbones and identical hydrophobic packing motifs so that the only difference was the placement and composition of hydrogen-bond networks. The designs were based on two trimers originating from the same superhelical parameters, but with unique hydrogen bond networks designated as “A” and “B”. Interface with only nonpolar residues are designated “X”. The three-letter code A, B, X was used combinatorially to generate new designs by placing A, B, or X at each of the four repeating cross sections of the supercoil. Five of the six combinatorial designs synthesized were found to be folded, thermostable, and assembled to form trimers. These five along with the two parent designs AAXX and XXBB and the all-hydrophobic control XXXX were crossed in an all-by-all array in a yeast two-hybrid binding experiment, which uses the expression of a reporter gene to measure how specifically two protein domains bind to each other. The results showed the hydrophobic domains to be promiscuous while the hydrogen bond networks mediate specificity.
Having demonstrated that their program HBNet provides a general computational method to accurately design hydrogen-bond networks, the authors claim that the ability to preorganize polar contacts without buried unsatisfied polar atoms should be broadly useful for enzyme design, small molecule binding, and matching polar protein interfaces.
The authors further propose that their two-ring structures provide a new class of protein oligomers with potential for programmable interactions analogous to Watson-Crick base pairing. Although Watson-Crick base pairing is largely limited to the antiparallel double helix, they propose that their designed protein hydrogen-bond networks allow specification of two-ring structures with a range of oligomerization states and supercoil geometries.
It should now become possible to develop new protein-based materials with the advantages of both polymers: DNA-like programmability and tunable specificity coupled with the geometric variability, interaction diversity, and catalytic function intrinsic to proteins.
Considering the striking progress the past decade has seen with both DNA nanotechnology and protein design, it should be especially interesting to watch how this proposal plays out.
—James Lewis, PhD
Discuss these news stories on Foresight’s Facebook page or on our Facebook group.