
Continuing yesterday’s discussion of two complementary approaches to balance designing protein structures with novel functions with designing protein structures with maximum stability, we focus on a method to create novel proteins by stitching together pieces of existing proteins, developed by Brian Kuhlman, one of two co-winners of the 2004 Feynman Prize, Theory category, and his collaborators. From the University of North Carolina Medical School newsroom “Scientists digitally mimic evolution to create new proteins“:
… researchers at the University of North Carolina School of Medicine have developed a method that creates novel proteins by stitching together pieces of already existing proteins.
The technique, called SEWING, is inspired by natural evolutionary mechanisms that also recombine portions of known proteins to produce new structures and functions. This approach can generate a diverse set of protein structures with many of the distinctive features that proteins require to carry out specific biological functions.
The findings, published today in the journal Science [“Design of structurally distinct proteins using strategies inspired by evolution” journal abstract, HHS Public Access author manuscript], could enable researchers to design proteins to play a variety of different roles in human biology and disease, such roles as catalysts, biosensors, and therapeutics.
“We can now begin to think about engineering proteins to do things that nothing else is capable of doing,” said senior study author Brian Kuhlman, PhD, professor of biochemistry and biophysics, and member of the UNC Lineberger Comprehensive Cancer Center. “The structure of a protein determines its function, so if we are going to learn how to design new functions, we have to learn how to design new structures. Our study is a critical step in that direction and provides tools for creating proteins that haven’t been seen before in nature.”
At the chemical level, proteins are composed of long chains of hundreds to thousands of subunits called amino acids – the building blocks of life. The sequence of these amino acids ultimately determines each protein’s unique geometry. Some sections of a protein might be folded back and forth onto itself like a paper fan; others might be coiled tightly like a spring. In all, scientists estimate that the human body contains about 100,000 different proteins, each the result of millions of years of evolutionary shuffling, culminating in a precise lineup of pleats, coils, and furrows required to carry out a specific job in the cell.
Traditionally, researchers have used computational protein design to recreate in the laboratory what already exists in the natural world. But in recent years, their focus has shifted toward inventing novel proteins with new functionality. These design projects all start with a specific structural “blueprint” in mind, and as a result are limited. Kuhlman and his colleagues believe that by removing the limitations of a pre-determined blueprint and taking cues from evolution they can more easily create functional proteins.
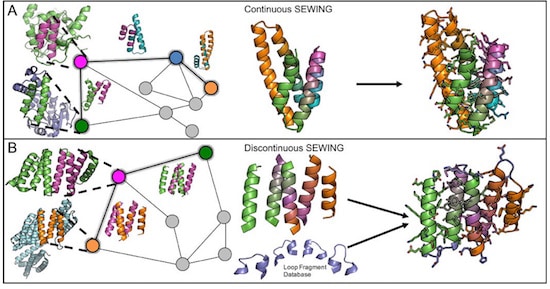
To mimic the mechanisms of natural protein evolution, they developed a computer design strategy called SEWING (Structure Extension With Native-substructure Graphs). First, the researchers took a slew of naturally occurring proteins and digitally chopped them up into well-defined pieces, as if turning a bunch of rag dolls into a pile of arms, legs, and heads. Then they performed a series of computational tests to figure out which pieces would fit well together. In nature, this step would involve looking for stretches of amino acid sequences that are similar between proteins. On the computer, it involved searching for regions of structural similarity so that – in the analogy of the rag doll – a hand would end up being stitched to an arm and then a shoulder, and not a head or a hip.
First author Tim M. Jacobs, PhD, a former graduate student in the Kuhlman lab, used this method to map out 50,000 of these stitched together proteins on the computer. He then tapped a number of different metrics to whittle down the list to the top 21 proteins, which he produced in the lab. Jacobs and colleagues took pictures of these proteins using x-ray crystallography and NMR, and found that the proteins contained all the unique structural varieties they had designed on the computer.
“We were excited that some had clefts or grooves on the surface, regions that naturally occurring proteins use for binding other proteins,” said Jacobs. “That’s important because if we wanted to create a protein that can act as a biosensor to detect a certain metabolite in the body, either for diagnostic or research purposes, it would need to have these grooves. Likewise, if we wanted to develop novel therapeutics, they would also need to attach to specific proteins.” …
Jacobs et al. propose that traditional efforts in protein design to produce idealized protein structures “may not always be the most effective starting points for engineering novel protein functions. Functional sites in proteins are often created from non-ideal structural elements, such as kinks, pockets and bulges.” Further, protein design methods begin with an idealized target structure in mind, while natural evolution depends on fitness provided by the evolved protein function, rather than predetermined structure. Their design strategy, called SEWING (Structure Extension With Native-substructure Graphs), builds new protein structures from small pieces of naturally occurring protein domains. They chose to extract two different types of substructures: the first, continuous stretches encompassing two secondary structure elements separated by a loop, to capture relative orientation and local packing interactions; the second, groups of 3-5 secondary structural elements that all make van der Waals contacts with each other but are not necessarily continuous in primary sequence, to maintain longer range tertiary interactions that are often conserved during protein evolution. A total of 33,928 continuous substructures and 4,584 discontinuous substructures were extracted from the protein data bank.
The above elements are combined and modified to develop new tertiary structures. Potential combinations are computationally tested to ensure structural fit, but without any target structure being required. This process produced about 7×1016 backbone structures for subsequent consideration. From this large number of possibilities 11 designs based on continuous SEWING and 10 designs based on discontinuous SEWING were selected for experimental characterization. Eight of the 11 continuous designs were soluble and readily purified. Two of these were hyperthermophiles, and one (CA01) exhibited an estimated melting temperature of 126 °C, with a crystal structure that exhibited excellent agreement with the design model, to an alpha-carbon root mean square deviation of only 80 pm.
Two of the 10 discontinuous SEWING were expressed well enough for purification. One of these, DA03, exhibited high thermostability. The other discontinuous design, DA05, did not readily crystallize, but NMR spectroscopy confirmed the presence of 4 of 5 of the designed helices, while the 5th was disordered. Redesigning that region using the continuous SEWING method yielded a new protein that adopts the designed conformation. The additional step of loop-building necessary for discontinuous SEWING may have accounted for the lower success using that method.
The diversity of models generated by SEWING is demonstrated, the authors claim, by the inclusion of kinked and curved helices, cavities and clefts, and a large range of helix-crossing angles. They note that the topologies of SEWING models is greater than seen with previously designed alpha-helical proteins, “which are restricted to coiled-coils, repeat proteins and up-down four helix bundles” The authors expect the diversity of SEWING designs to further increase when alternative substructures are included, such as β-α motifs and β-hairpins.
We anticipate that this structural diversity will be advantageous for functional design, as every backbone generated with SEWING has new surface and pocket features that provide potential binding sites for ligands or macromolecules. Additionally, SEWING offers an approach for stitching together functional motifs from naturally occurring proteins, an evolutionary mechanism to generate multi-functional proteins and allosteric systems.
The study described in our most recent post and this study together demonstrate substantial control over biomolecular shape and interactions, complementing previous accomplishments in designing protein stability and extending protein design space. We can hope that increasing control over the balance between stabilizing and functional features will lead to designing new protein functions, and eventually more complex and capable molecular machine systems.
—James Lewis, PhD
Discuss these news stories on Foresight’s Facebook page or on our Facebook group.