
Regular readers will have noticed that the de novo design of proteins not found in nature has become an increasingly active area of nanotechnology research the past several years, including eight advances this past year that we have cited (here, here, here, here, here, here, here, here). To put this rapid acceleration of progress in perspective, David Baker’s group, source of 7 of the above 8 advances, recently published (Sept, 2016) a review “The coming of age of de novo protein design“:
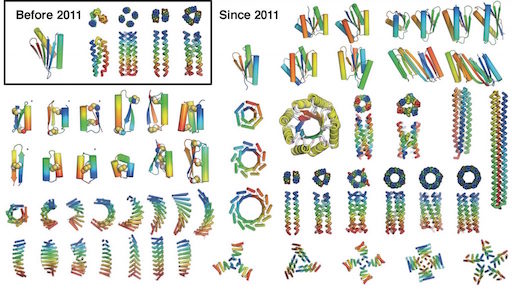
Most protein design efforts to date have focused on reengineering existing proteins found in nature. By contrast, de novo protein design generates new structures from scratch, with sequences unrelated to naturally occurring proteins. Before 2011, the only successful de novo designed proteins were Top7 (2003), and an array of coiled coil peptides (helical bundles). In the past five years, the field of de novo protein design has exploded. The wealth of new structures, and advancements in methodology, should now now allow proteins to be precisely crafted and custom-made to solve specific modern-day problems.
The review is published in Nature (journal abstract) and the full text PDF is available courtesy of the Baker lab. The journal abstract:
There are 20200 possible amino-acid sequences for a 200-residue protein, of which the natural evolutionary process has sampled only an infinitesimal subset. De novo protein design explores the full sequence space, guided by the physical principles that underlie protein folding. Computational methodology has advanced to the point that a wide range of structures can be designed from scratch with atomic-level accuracy. Almost all protein engineering so far has involved the modification of naturally occurring proteins; it should now be possible to design new functional proteins from the ground up to tackle current challenges in biomedicine and nanotechnology.
The authors cite the diverse functions that natural proteins have evolved to execute through several hundred million years of random variation and selective pressure working on primordial proteins: using solar energy to manufacture complex molecules, ultrasensitive detection of small molecules and light, the conversion of pH gradients into chemical bonds, and transforming chemical energy into work. They estimate the total number of distinct proteins produced by extant organisms as 1012, an extremely tiny fraction of the 20200 (equals approximately 1.6 x 10260) distinct sequences possible for a 200-residue protein, indicating that evolution has explored only a tiny region of the sequence space available to proteins. Because evolution proceeds by incremental variation and selection, natural proteins are not spread uniformly across sequence space. Thus the vast spaces not sampled by evolution comprise the arena for de novo protein design based on the principles of protein biophysics, specifically that proteins fold into the lowest energy states accessible to their amino acid sequences.
The review describes the physical basis of the energy function used for design calculations, and the approaches used to overcome the problem of sampling an immense sequence space. This review is based on the authors’ experience with the Rosetta structure prediction and design methodology that they have developed; other de novo protein design software described elsewhere is cited. The authors attribute recent progress not only to advances in understanding and computer methods, but also to a steady increase in computing power, and a dramatic improvement in DNA synthesis methods, greatly lowering the cost of the synthetic genes needed to express designed proteins in bacteria. With respect to lower prices for synthetic DNA, it is interesting to note that a technical advance that accelerates one path to advanced nanotechnology (DNA nanotechnology) can also independently advance a different path (de novo protein design).
The physical principles that underlie protein design are described.
- The force driving protein folding is the burial of hydrophobic residues in the protein’s core, away from solvent, with side chains packed as closely as possible without unfavorable atomic overlaps.
- Polar groups that become buried upon folding must form intra-protein hydrogen bonds.
- Steric and torsional effects favor certain backbone geometries and disfavor others.
The authors first consider the ab initio structure prediction problem: finding the lowest energy structure for a fixed amino acid sequence in the absence of information about the structures of evolutionarily related proteins. Because the size of the backbone conformational space is huge, predicting structure without information on related proteins is only possible for the smallest proteins. In general, successful structure prediction requires a number of distance constraints from a set of proteins that have co-evolved.
Because only a finite number of backbones can be sampled computationally, sequence-independent constraints on backbone geometry must be used. For example, polar atoms of the backbone with make hydrogen bonds within the chain in α-helices or β-sheets, or contact the solvent in exposed loops. Much de novo protein design work emphasizes designing ideal proptein structures with unkinked α-helices and β-strands. By contrast, most natural proteins contain irregular features, which reduce the free energy of folding, that arise either from selection for function or from neutral drift. The authors note that a free energy of folding of 8 kcal per mole would suffice to insure a folded population of more than 99.999%, so that evolution would have been under little pressure to optimize folding beyond that.
The procedure is described that was used to design a wide range of ideal αβ protein structures, including Top7, the first (2003) globular protein to be designed with a fold not observed in nature. Modifications and additions to the procedure are described, including designing proteins with internal symmetry in which a single idealized unit is repeated numerous times. such as toroids and barrels. The sequences that have been designed, expressed, and characterized are often exceptionally stable and differ greatly from those found in nature, suggesting that naturally occurring proteins sample only a tiny fraction of stable protein structures that can be realized. Some of the examples given in this review are among the eight advances cited at the top of this post.
The authors cite some of the many accomplishments that have resulted from computational protein-design efforts that have engineered new functions on scaffolds derived from natural proteins, and propose that the capabilities of next generation designed functional proteins could greatly exceed those of first-generation designed proteins based on native scaffolds. They further point to opportunities presented by incorporating new chemistries and unnatural amino acids {in the near future we will cover their work that they cite here on hyperstable constrained peptides}.
Addressing the limitations of protein design as currently practiced, they acknowledge that only a faction of protein designs adopt stable folded structures when produced in the bacterium E. coli. Failure is often due to lack of solubility and formation of unintended oligomers, both probably arising from unanticipated intermolecular hydrophobic interactions. The authors expect that it should become increasingly possible to identify the factors that differ between soluble and insoluble designs. They further note that their highest success rates have been with peptides that were synthesized chemically, so that part of the reason for failures might lie with toxicity of certain designs expressed in E. coli, or with complexities of the bacterium’s biology. Also, high successes with α-helical repeat designs indicates sequence repetition is probably favored over alternative structures.
The review finishes with a consideration of challenges and opportunities of ongoing work in de novo protein design. First, they note a fundamental problem encountered when redesigning natural proteins for new functions, such as new catalytic activities, altering many residues to introduce a new function can inadvertently alter the structure, such as by introducing unanticipated loops. Since native proteins are often only marginally stable, changes can lead to unfolding or aggregation. In contrast, the very high stability of de novo designed proteins should render them more suitable as starting points for creating new functions.
Although de novo designed proteins begin with ideal structures, the introduction of functional sites and binding interfaces will “inevitably compromise this ideality” and thus render the structure less stable. Binding surfaces usually contain hydrophobic residues, and are thus more prone to aggregation, and the active sites of enzymes have some mobility to allow substrates to enter and products to leave. To address these challenges, the authors first suggest designing recessed cavities into proteins to enable ligand and substrate binding. They have already addressed this challenge in a paper they have just published, which we will feature in an upcoming post. Finally, the authors note that sophisticated functions like allostery and signalling found in natural proteins …
… emerge in protein systems with multiple low-energy states and moving parts that can be toggled by external stimuli. To achieve such capabilities, which could have widespread applications in the design of molecular machines to tackle problems ranging from tumour recognition to computing, will require proteins to be designed with multiple, distinct energy minima.
The review cites a study published two years ago that demonstrates the capability to design a functional protein with two alternative states.
Overcoming these challenges in the years ahead is an exciting prospect. Success would signal a technological advance that is analogous to the transition from the Stone Age to the Iron Age. Instead of building new proteins from those that already exist in nature, protein designers can now strive to precisely craft new molecules to solve specific problems — just as modern technology does outside of the realm of biology.
By providing an overview of progress along a major path to atomically precise nanotechnology, and a clear view of near-term challenges and how they might be met, this review contributes significantly to a roadmap from current molecular science to a future technology of productive nanosystems and general purpose, high throughput atomically precise manufacturing.
—James Lewis, PhD
Discuss these news stories on Foresight’s Facebook page or on our Facebook group.