We have pointed to examples of how atomically precise nanotechnology might open the road to developing quantum computers (Atomically precise location of dopants a step toward quantum computers, August 4th, 2016; Architecture for atomically precise quantum computer in silicon, November 9th, 2015; A nanotechnology route to quantum computers through hybrid rotaxanes, March 27th, 2009). The converse process of quantum computing facilitating the development of atomically precise nanotechnology by enabling quantum simulations too large to be tractable on a classical computer was noted by Richard Terra writing on this blog (Quantum computers for quantum physics calculations, July 5th, 2002). This converse process took a big step forward with an advance from IBM.
MIT Technology Review reports “IBM Has Used Its Quantum Computer to Simulate a Molecule—Here’s Why That’s Big News“
We just got a little closer to building a computer that can disrupt a large chunk of the chemistry world, and many other fields besides. A team of researchers at IBM have successfully used their quantum computer, IBM Q, to precisely simulate the molecular structure of beryllium hydride (BeH2). It’s the most complex molecule ever given the full quantum simulation treatment.
Molecular simulation is all about finding a compound’s ground state—its most stable configuration. Sounds easy enough, especially for a little-old three-atom molecule like BeH2. But in order to really know a molecule’s ground state, you have to simulate how each electron in each atom will interact with all of the other atoms’ nuclei, including the strange quantum effects that occur on such small scales. This is a problem that becomes exponentially harder as the size of the molecule increases. …
The IBM team published in Nature a new algorithm to calculate the ground state of BeH2 “Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets ” [abstract, full text preprint]. An IBM Research blog post by three of the seven authors of the Nature paper provides further explanation of the quantum algorithm they implemented with IBM’s seven qubit superconducting quantum hardware “How to measure a molecule’s energy using a quantum computer“:.
Simulating molecules on quantum computers just got much easier with IBM’s superconducting quantum hardware. In a recent research article published in Nature, … we implement a new quantum algorithm capable of efficiently computing the lowest energy state of small molecules. By mapping the electronic structure of molecular orbitals onto a subset of our purpose-built seven qubit quantum processor, we studied molecules previously unexplored with quantum computers, including lithium hydride (LiH) and beryllium hydride (BeH2). The particular encoding from orbitals to qubits studied in this work can be used to simplify simulations of even larger molecule and we expect the opportunity to explore such larger simulations in the future, when the quantum computational power (or “quantum volume”) of IBM Q systems has increased.
While BeH2 is the largest molecule ever simulated by a quantum computer to date, the considered model of the molecule itself is still simple enough for classical computers to simulate exactly. This made it a test case to push the limits of what our seven qubit processor could achieve, further our understanding of the requirements to enhance the accuracy of our quantum simulations, and lay the foundational elements necessary for exploring such molecular energy studies.
The best simulations of molecules today are run on classical computers that use complex approximate methods to estimate the lowest energy of a molecular Hamiltonian. A “Hamiltonian” is a quantum mechanical energy operator that describes the interactions between all the electron orbitals* and nuclei of the constituent atoms. The “lowest energy” state of the molecular Hamiltonian dictates the structure of the molecule and how it will interact with other molecules. Such information is critical for chemists to design new molecules, reactions, and chemical processes for industrial applications.…
Although our seven qubit quantum processor is not fully error-corrected and fault-tolerant, the coherence times of the individual qubits last about 50 µs. It is thus really important to use a very efficient quantum algorithm to make the most out of our precious quantum coherence and try to understand molecular structures. The algorithm has to be efficient in terms of number of qubits used and number of quantum operations performed.
Our scheme contrasts from previously studied quantum simulation algorithms, which focus on adapting classical molecular simulation schemes to quantum hardware – and in so doing not effectively taking into account the limited overheads of current realistic quantum devices.
So, instead of forcing classical computing methods onto quantum hardware, we have reversed the approach and asked: how can we extract the maximal quantum computational power out of our seven qubit processor?
Our answer to this combines a number of hardware-efficient techniques to attack the problem:
- First, a molecule’s fermionic Hamiltonian is transformed into a qubit Hamiltonian, with a new efficient mapping that reduces the number of qubits required in the simulation.
- A hardware-efficient quantum circuit that utilizes the naturally available gate operations in the quantum processor is used to prepare trial ground states of the Hamiltonian.
- The quantum processor is driven to the trial ground state, and measurements are performed that allow us to evaluate the energy of the prepared trial state.
- The measured energy values are fed to a classical optimization routine that generates the next quantum circuit to drive the quantum processor to, in order to further reduce the energy.
- Iterations are performed until the lowest energy is obtained to the desired accuracy.
With future quantum processors, that will have more quantum volume, we will be able to explore the power of this approach to quantum simulation for increasingly complex molecules that are beyond classical computing capabilities. The ability to simulate chemical reactions accurately, is conductive to the efforts of discovering new drugs, fertilizers, even new sustainable energy sources.
The experiments we detail in our paper were not run on our currently publically available five qubit and 16 qubit processors on the cloud. But developers and users of the IBM Q experience can now access quantum chemistry Jupyter notebooks on the QISKit github repo. On the five qubit system, users can explore ground state energy simulation for the small molecules hydrogen and LiH. Notebooks for larger molecules are available for those with beta access to the upgraded 16-qubit processor.
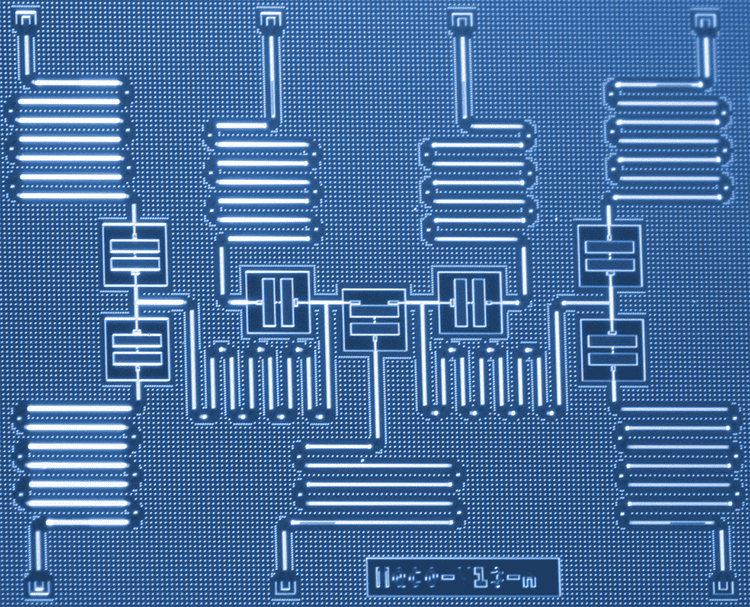
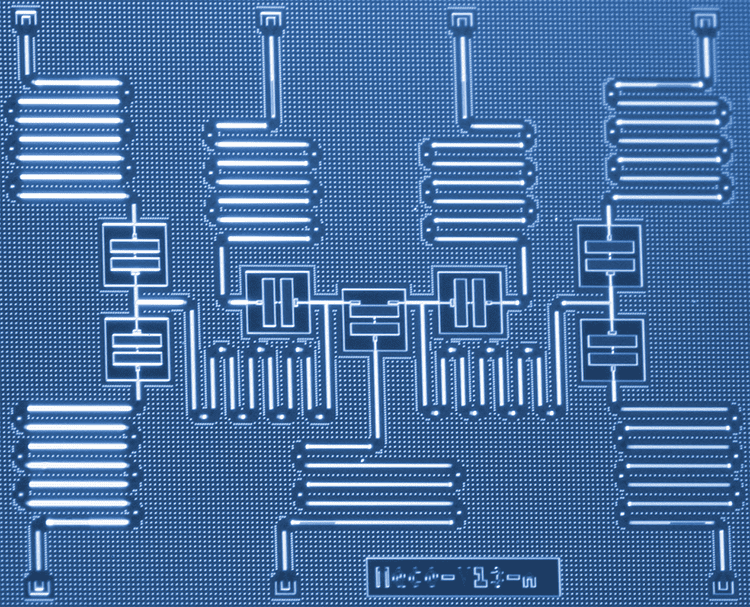
Information about the physical implementation if IBM’s superconducting quantum hardware and qubits can be found on IBM Q experience under Frequently Asked Questions:
What is the qubit that you are physically using?
The qubit we use is a fixed-frequency superconducting transmon qubit. It is a Josephson-junction-based qubit that is insensitive to charge noise. For more information on this type of qubit please see here (Koch et al. 2007, abstract, arxiv preprint). We use fixed-frequency qubits, as opposed to tunable qubits, to minimize our sensitivity to external magnetic field fluctuations that could corrupt the quantum information.
How do you make the qubits?
The superconducting qubits are fabricated at IBM. The devices are made on silicon wafers with superconducting metals such as niobium and aluminum. Details about the fabrication processes are given in these references (Chow et al. 2014, Córcoles et al. 2015).
The qubits used in the work described here appear to be fabricated using optical lithography, so are far from requiring atomically precise fabrication technology. On the other hand, quantum algorithm advances as exemplified here may facilitate the computational analysis and design of general purpose high-throughput atomically precise manufacturing systems, as well as systems to implement artificial general intelligence. The opportunities for synergy among these emerging, world-shaping technologies would appear to be large and growing.
—James Lewis, PhD