Protein design has been one of the major paths from current fabrication technology toward the goal of general purpose, high-throughput atomically precise manufacturing since Foresight co-founder Eric Drexler proposed it in 1981. It also produced some of the earliest promising results. Although de novo protein design was at first slow, progress has accelerated since David Baker (University of Washington) and Brian Kuhlman (University of North Carolina) won the 2004 Foresight Feynman Prize for Theoretical work for the creation of the RosettaDesign software for modeling and analysis of protein structures. Among recent successes: “From de novo protein design to molecular machine systems“, “Designing novel protein backbones through digital evolution“, and “Rational design of protein architectures not found in nature“. Another milestone accomplished the design of new backbone structures to fit into target binding, and opened up previously inaccessible regions of shape space to design and fabricate new parts for complex molecular machine systems. A September, 2016 news release from the Baker Lab “Accurate de novo design of hyperstable constrained peptides“:
Small constrained peptides combine the stability of small molecule drugs with the selectivity and potency of antibody-based therapeutics. However, peptide-based therapeutics have largely remained underexplored due to the limited diversity of naturally occurring peptide scaffolds, and a lack of methods to design them rationally.
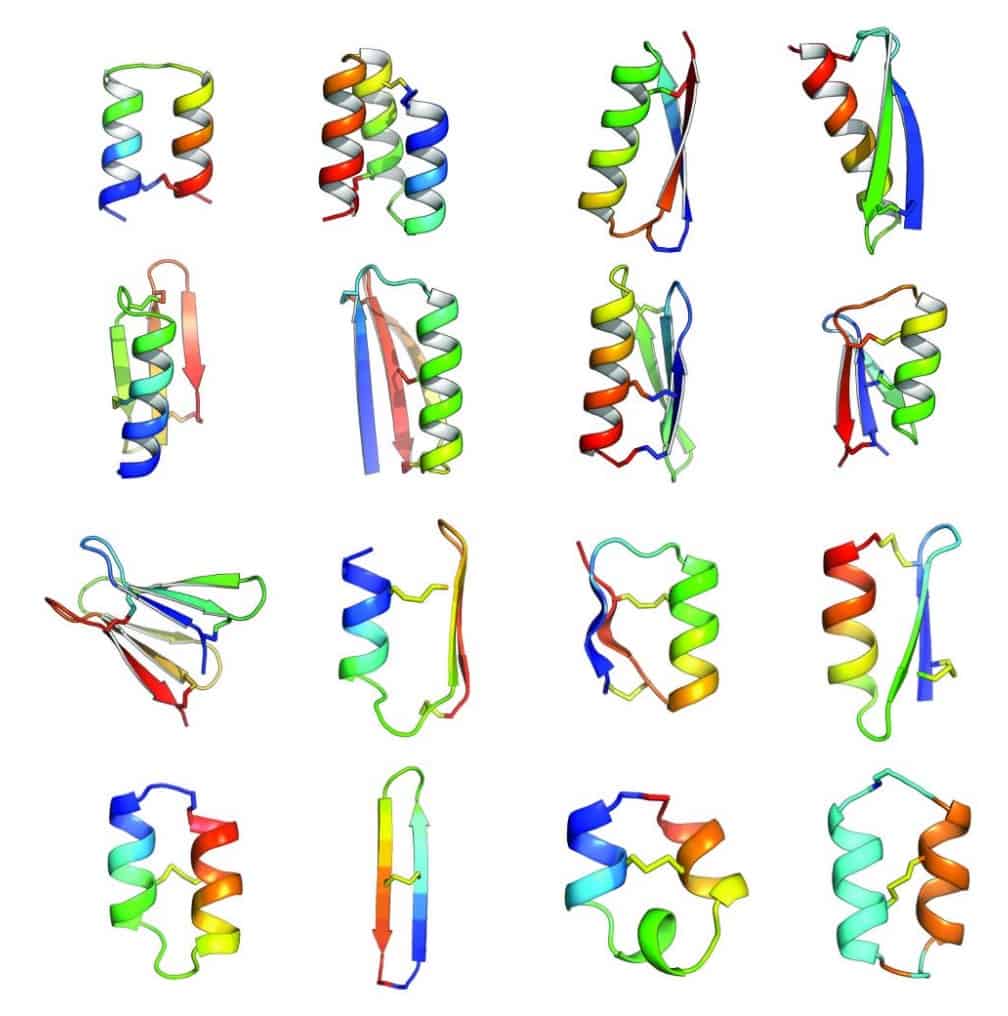
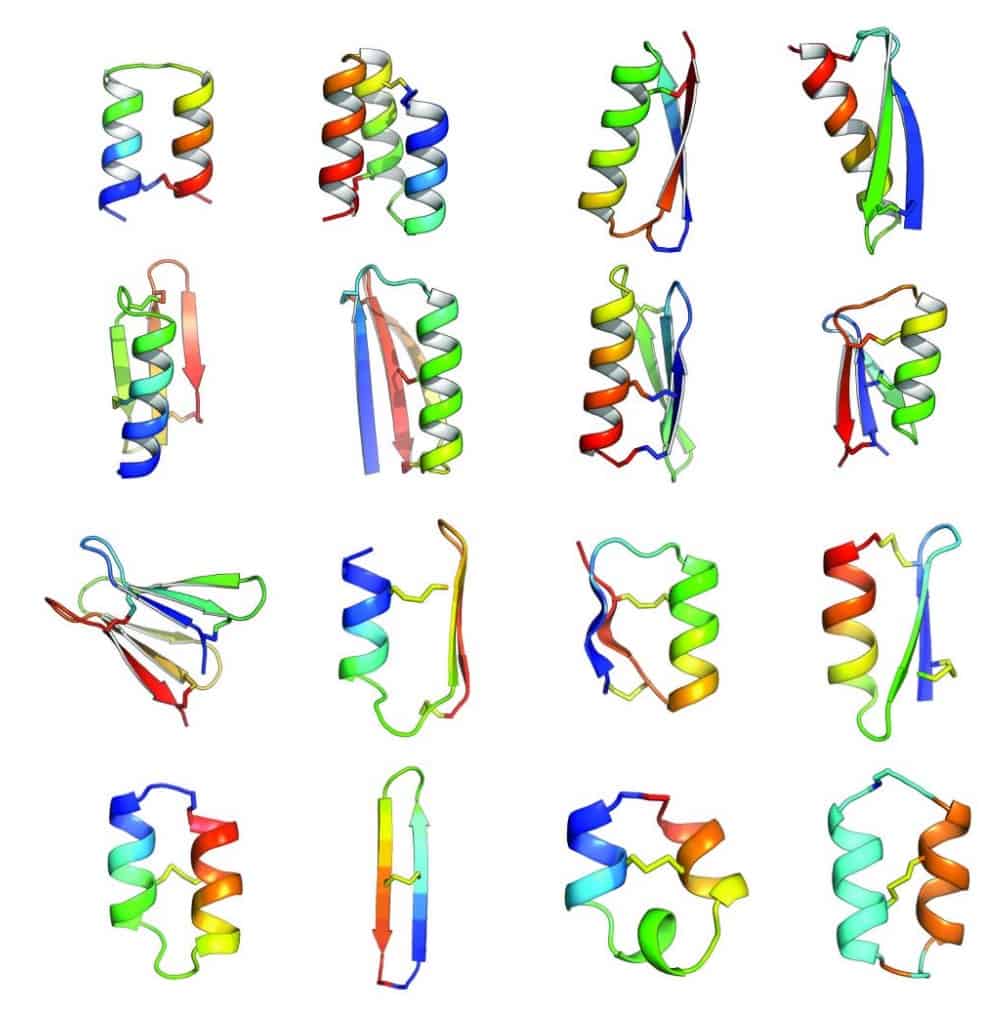
In an article published in Nature this week [abstract, PDF courtesy of Baker lab], Baker lab scientists and collaborators describe the development of computational methods for de novo design of constrained peptides with exceptional stabilities. They used these computational methods to design 18-47 residue constrained peptides with diverse shapes and sizes. The designed peptides presented in the paper cover three broad categories: 1) genetically encodable disulfide cross-linked peptides, 2) synthetic disulfide cross-linked peptides with non-canonical sequences, and 3) cyclic peptides with non-canonical backbones and sequences. Experimentally determined structures for these peptides are nearly identical to their design models.
By including D-amino acids (mirror images of the L-amino acids), and thus expanding the palette of building blocks, Baker lab scientists designed peptides in a sequence and structure space sampled rarely by Nature. Indeed, the article describes successful design of a cyclic 2-helix peptide of mix chirality that represents a shape beyond natural secondary- and tertiary structure.
These designed peptides also exhibit exceptional stability to thermal and chemical denaturation, and thus could serve as attractive scaffolds for design of novel peptide-based therapeutics. More broadly, development of this new computational toolkit to precisely design constrained peptides opens the door for “on-demand” development of a new generation of peptide-based therapeutics.
This research begins with the observation that constrained peptides are an unexplored frontier for drug discovery that is made interesting by the fact that among the small number of examples known are some of the most potent pharmacologically active compounds known. these peptides are constrained by disulfide bonds or backbone cyclization to favor conformations that precisely complement their targets. The inability to achieve global shape complementarity with targets reveals the need for a method to create constrained peptides that provide precise control over the size and shape of the designed molecules. The desire of the researchers to access “broad regions of peptide structure and function space not explored by evolution” provides a motivation to incorporate non-canonical backbones and unnatural amino acids.
Of course, the computational design of covalently constrained peptides with new strutures and non-canonical backbones presents new challenges, including mixed chirality. The Rosetta software suite was used for all of the design calculations in this article. A diverse array of 18-47 residue peptides was designed. These included two classes of peptides: (1) genetically encodable (i.e., using only the 20 amino acids specified by the universal genetic code, often called the canonical amino acids) disulfide-rich peptides, (2) heterochiral peptides with non-canonical sequences. The authors note that genetic encodability has the advantage of compatibility with high-throughput selection methods like phage, ribosome, and yeast display, while incorporation of non-canonical components opens access to new types of structures. For the former class, they selected nine combinations of α-helices and β-strands. The latter class included α-helices and β-strands connected by loop segments containing D-amino acid residues, non-canonical amino acids, and cyclic structures.
Genetically encodable disulfide-constrained peptides
For the nine chosen topologies of genetically encodable disulfide-constrained peptides, Monte Carlo-based assembly of short protein fragments was used to construct backbone conformations, which were then scanned for sites capable of hosting disulfide bonds with nearly ideal geometry. One ot three disulfides bonds were incorporated and low energy sequences were designed and optimized using the Rosetta all-atom force field. Rosetta ab initio structure prediction calculations were carried out for each designed sequence, resulting in a diverse set of 130 designs for which the target structure was in a deep global free-energy minimum (i.e., the structure would be very stable). Genes were constructed for each design and expressed in the bacterium Escherichia coli or in cultured mammalian cells. Since disulfide bonds would be unlikely to form in the reducing environment of the cytoplasm, gene expression was engineered to secrete the designed proteins, which were analyzed for signs that the disulfide bond had formed consistent with the designed topology. 29 designs passed this test, and one representative design was chosen from each of the nine topologies for further biochemical characterization.
One of the nine designs produced a protein that could be crystallized. The structure was determined to a resolution of 0.209 nm. The details of the structure were in excellent agreement with the design model. The protein was thermostable and completely resistant to chemical denaturation.
The eight designs that could not be crystallized were expressed as isotopically labelled peptides and the structures determined by nuclear magnetic resonance (NMR) spectroscopy. The formation of the designed disulfide bonds was confirmed. “Taken together, the X-ray crystallographic and NMR structures demonstrate that our computational approach enables accurate design of protein main-chain conformation, disulfide bonds and core residue rotamers.”
Synthetic heterochiral disulfide-constrained peptides
To design shorter disulfide-constrained peptides incorporating
both l- and d-amino acids, the rosetta energy function was generalized to support D-amino acids and mixed chirality designs. Since chemical synthesis required to synthesize peptides that cannot be genetically encoded is laborious, automated computational screening techniques were developed to supplement Rosetta ab initio screening with molecular dynamics (MD) evaluation. Sequences were designed favoring D-amino acids at positions with positive main chain φ dihedral angle values. A single low energy design was selected for each of three topologies evaluated, chemically synthesized, and structurally characterized by NMR. All three gave NMR spectra consistent with the secondary structure of the design. High resolution NMR solution structures showed close agreement for two of the designs. The third differed from the design model by having an unwound carboxyl terminus, but a second design chosen for that topology had a structure very close to the design model. All three designs were very thermostable.
Synthetic backbone-cyclized peptides
A generalized kinematic loop closure method (named GenKIC) was implemented to samo arbitrary covalently linked atom chains capable of connecting the termini. Each GenKIC chain-closure attempt involved perturbing multiple chain degrees of freedom, then enforcing loop closure with ideal peptide bond geometry. “Sequence design, backbone relaxation, and in silico structure validation using MD simulation and Rosetta ab initio structure prediction were carried out with terminal bond geometry constraints”. Cyclic peptides were synthesized for three topologies, and their structures determined with NMR spectroscopy. All three peptides had structures very close to their design models, and all three were extremely stable to thermal denaturation and resistant to chemical denaturation. They were exceptionally stable given their small sizes.
Beyond natural secondary and tertiary structure
A “heterochiral, backbone-cyclized, two-helix topology with
one non-canonical left-handed α-helix and one canonical right-handed
α-helix” provided a final test of the design methodology. For validation by ab initio structure prediction, it was necessary to develop a new, GenKIC-based protocol since the standard Rosetta method uses uses fragments of native proteins, which typically do not contain left-handed helices. The selected design for this topology is a 26-residu protein with one D-cysteine,L-cysteine disulfide bond connecting the right-handed and left-handed α-helices. There was an excellent match between the NMR structure ensemble and the design model. This success demonstrated that the authors’ computational methods are general enough to design in a conformational space not explored by nature.
The authors point out that of the sixteen constrained peptide topologies designed, the twelve for which the strutures were experimentally determined were in close agreement with the design models. Unlike the natural constrained peptide families, these designed peptides are not limited to particular sizes, shapes, or disulfide connectivities.
Here we have focused on extending sampling and scoring methods to permit design with d-amino acids and cyclic backbones, but the new tools are fully generalizable to peptides containing more exotic building-blocks, such as amino acids with non-canonical sidechains or non-canonical backbones.
This research was clearly focused on extending de novo peptide design methods to provide a greater variety of protein components for drug discovery and therapeutic applications. Drugs and biotech therapies, whether small molecules or protein or other biomolecules, are all molecules sought to enhance or alter the functions of the complex natural molecular machine systems that comprise cells and organisms. Other complex molecular machine systems, as yet not designed, will play crucial roles along the paths to productive nanosystems and general purpose, high throughput atomically precise manufacturing.
—James Lewis, PhD
Discuss these news stories on Foresight’s Facebook page or on our Facebook group.